
Перенос молекулярной динамики на CUDA. Часть II: Суммирование по Эвальду
где q – заряд частицы, rij – расстояние между частицами, С – некоторая постоянная, зависящая от выбора единиц измерения. В системе СИ это — , в СГС — 1, в моей программе (где энергия выражена в электронвольтах, расстояние в ангстремах, а заряд в элементарных зарядах) C примерно равно 14.3996.
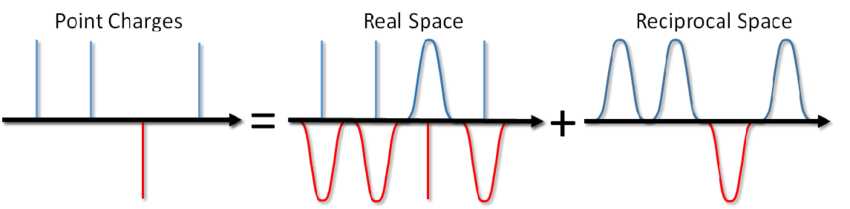
Ну и что, скажете вы? Просто добавим соответствующее слагаемое в парный потенциал и готово. Однако, чаще всего в МД моделировании используют периодические граничные условия, т.е. моделируемая система со всех сторон окружена бесконечным количеством её виртуальных копий. В этом случае каждый виртуальный образ нашей системы будет взаимодействовать со всеми заряженными частицами внутри системы по закону Кулона. А поскольку Кулоновское взаимодействие убывает с расстоянием очень слабо (как 1/r), то отмахнуться от него так просто нельзя, сказав, что с такого-то расстояния мы его не вычисляем. Ряд вида 1/x расходится, т.е. его сумма, в принципе, может расти до бесконечности. И что же теперь, миску супа не солить? Убьёт электричеством?
…можно не только солить суп, но и посчитать энергию Кулоновского взаимодействия в периодических граничных условиях. Такой метод был предложен Эвальдом ещё в 1921 году для расчета энергии ионного кристалла (можно также посмотреть в википедии). Суть метода заключатся в экранировании точечных зарядов и последующим вычетом функции экранирования. При этом часть электростатического взаимодействия сводится к короткойдействующему и его можно просто обрезать стандартным способом. Оставшаяся дальнодействующая часть эффективно суммируется в пространстве Фурье. Опуская вывод, который можно посмотреть в статье Блинова или в той же книге Френкеля и Смита сразу запишу решение, называемое суммой Эвальда:
где α – параметр, регулирующий соотношение вычислений в прямом и обратном пространствах, k – вектора в обратном пространстве по которым идёт суммирование, V – объём системы (в прямом пространстве). Первая часть (Ereal) является короткодействующей и вычисляется в том же цикле, что и другие парные потенциалы, смотри функцию real_ewald в предыдущей статье. Последний вклад (Eсonst) является поправкой на самовзаимодействие и часто называется «постоянной частью», поскольку не зависит от координат частиц. Её вычисление тривиально, поэтому мы остановимся только на второй части Эвальдовой суммы (Erec), суммировании в обратном пространстве. Естественно, во времена вывода Эвальда молекулярной динамики не было, кто впервые использовал этот метод в МД найти мне не удалось. Сейчас любая книга по МД содержит его изложение как некий золотой стандарт. К книге Аллена даже прилагается пример кода на фортране. К счастью, у меня остался код, написанный когда-то на С для последовательной версии, осталось только его распараллелить (я позволил себе опустить некоторые объявления переменных и другие несущественные детали):
void ewald_rec()
{
int mmin = 0;
int nmin = 1;
// массивы где хранятся iexp(x[i] * kx[l]),
double** elc;
double** els;
//... iexp(y[i] * ky[m]) и
double** emc;
double** ems;
//... iexp(z[i] * kz[n]),
double** enc;
double** ens;
// временные массивы для произведений iexp(x*kx)*iexp(y*ky)
double* lmc;
double* lms;
// и для q[i] * iexp(x*kx)*iexp(y*ky)*iexp(z*kz)
double* ckc;
double* cks;
// ПРЕДВАРИТЕЛЬНОЕ ЗАПОЛНЕНИЕ МАССИВОВ
eng = 0.0;
for (i = 0; i < Nat; i++) // цикл по атомам
{
// emc/s[i][0] и enc/s[i][0] уже заполнены на этапе инициализации
// в массив elc/s нужно обновить, см. далее
elc[i][0] = 1.0;
els[i][0] = 0.0;
// iexp(kr)
sincos(twopi * xs[i] * ra, els[i][1], elc[i][1]);
sincos(twopi * ys[i] * rb, ems[i][1], emc[i][1]);
sincos(twopi * zs[i] * rc, ens[i][1], enc[i][1]);
}
// заполняем следующие элементы массива emc/s[i][l] = iexp(y[i]*ky[l]) итеративно, используя комплексное умножение
for (l = 2; l < ky; l++)
for (i = 0; i < Nat; i++)
{
emc[i][l] = emc[i][l - 1] * emc[i][1] - ems[i][l - 1] * ems[i][1];
ems[i][l] = ems[i][l - 1] * emc[i][1] + emc[i][l - 1] * ems[i][1];
}
// заполняем следующие элементы массива enc/s[i][l] = iexp(z[i]*kz[l]) итеративно, используя комплексное умножение
for (l = 2; l < kz; l++)
for (i = 0; i < Nat; i++)
{
enc[i][l] = enc[i][l - 1] * enc[i][1] - ens[i][l - 1] * ens[i][1];
ens[i][l] = ens[i][l - 1] * enc[i][1] + enc[i][l - 1] * ens[i][1];
}
// ГЛАВНЫЙ ЦИКЛ ПО ВСЕМ K-ВЕКТОРАМ:
for (l = 0; l < kx; l++)
{
rkx = l * twopi * ra;
// записываем exp(ikx[l]) в ikx[0] для сохранения памяти
if (l == 1)
for (i = 0; i < Nat; i++)
{
elc[i][0] = elc[i][1];
els[i][0] = els[i][1];
}
else if (l > 1)
for (i = 0; i < Nat; i++)
{
// iexp(kx[0]) = iexp(kx[0]) * iexp(kx[1])
x = elc[i][0];
elc[i][0] = x * elc[i][1] - els[i][0] * els[i][1];
els[i][0] = els[i][0] * elc[i][1] + x * els[i][1];
}
for (m = mmin; m < ky; m++)
{
rky = m * twopi * rb;
// заполняем временный массив произведением iexp(kx*x[i]) * iexp(ky*y[i])
if (m >= 0)
for (i = 0; i < Nat; i++)
{
lmc[i] = elc[i][0] * emc[i][m] - els[i][0] * ems[i][m];
lms[i] = els[i][0] * emc[i][m] + ems[i][m] * elc[i][0];
}
else // для отрицательных значений m используем комплексное сопряжение:
for (i = 0; i < Nat; i++)
{
lmc[i] = elc[i][0] * emc[i][-m] + els[i][0] * ems[i][-m];
lms[i] = els[i][0] * emc[i][-m] - ems[i][-m] * elc[i][0];
}
for (n = nmin; n < kz; n++)
{
rkz = n * twopi * rc;
rk2 = rkx * rkx + rky * rky + rkz * rkz;
if (rk2 < rkcut2) // используем радиус обрезания
{
// вычисляем сумму (q[i]*iexp(kr[k]*r[i])) - зарядовую плотность
sumC = 0; sumS = 0;
if (n >= 0)
for (i = 0; i < Nat; i++)
{
//считываем заряд частицы
ch = charges[types[i]].charge;
ckc[i] = ch * (lmc[i] * enc[i][n] - lms[i] * ens[i][n]);
cks[i] = ch * (lms[i] * enc[i][n] + lmc[i] * ens[i][n]);
sumC += ckc[i];
sumS += cks[i];
}
else // для отрицательных индексов используем комплексное сопряжение:
for (i = 0; i < Nat; i++)
{
//считываем заряд частицы
ch = charges[types[i]].charge;
ckc[i] = ch * (lmc[i] * enc[i][-n] + lms[i] * ens[i][-n]);
cks[i] = ch * (lms[i] * enc[i][-n] - lmc[i] * ens[i][-n]);
sumC += ckc[i];
sumS += cks[i];
}
//наконец вычисляем энергию и силы
akk = exp(rk2 * elec->mr4a2) / rk2;
eng += akk * (sumC * sumC + sumS * sumS);
for (i = 0; i < Nat; i++)
{
x = akk * (cks[i] * sumC - ckc[i] * sumS) * C * twopi * 2 * rvol;
fxs[i] += rkx * x;
fys[i] += rky * x;
fzs[i] += rkz * x;
}
}
} // end n-loop (over kz-vectors)
nmin = 1 - kz;
} // end m-loop (over ky-vectors)
mmin = 1 - ky;
} // end l-loop (over kx-vectors)
engElec2 = eng * С * twopi * rvol;
}
Пара пояснений к коду: функция считает комплексную экспоненту (в комментариях к коду она обозначена iexp, чтобы убрать мнимую единицу из скобок) от векторного произведения k-вектора на радиус-вектор частицы для всех k-векторов и для всех частиц. Эта экспонента домножается на заряд частицы. Далее вычисляется сумма таких произведений по всем частицам (внутренняя сумма в формуле для Erec), у Френкеля она называется зарядовой плотностью, а у Блинова — структурным фактором. Ну а далее, на основании этих структурных факторов вычисляется энергия и силы, действующие на частицы. Компоненты k-векторов (2π*l/a, 2π*m/b, 2π*n/c) характеризуются тройкой целых чисел l, m и n, которые и пробегаются в циклах до заданных пользователем пределов. Параметры a, b и c – это размеры моделируемой системы в измерениях x, y и z соответственно (вывод верен для системы с геометрией прямоугольного параллелепипеда). В коде 1/a, 1/b и 1/с соответствуют переменным ra, rb и rc. Массивы под каждую величину представлены в двух экземплярах: под действительную и мнимую части. Каждый следующий k-вектор в одном измерении получается итеративно из предыдущего путем комплексного умножения предыдущего на единичный, чтобы каждый раз не считать синус с косинусом. Массивы emc/s и enc/s заполняются для всех m и n, соответственно, а массив elc/s значение для каждого l>1 помещает в нулевой индекс по l в целях экономии памяти.
В целях распараллеливания выгодно «вывернуть» порядок циклов так, чтобы внешний цикл пробегался по частицам. И тут мы видим проблему – распараллелить эту функцию можно только до вычисления суммы по всем частицам (зарядовой плотности). Дальнейшие вычисления опираются на эту сумму, а она будет рассчитана только когда все потоки закончат работу, поэтому придётся разбить эту функцию на две. Первая вычисляет считает зарядовую плотность, а вторая – энергию и силы. Замечу, что во второй функции снова потребуется величина qiiexp(kr) для каждой частицы и для каждого k-вектора, вычисленная на предыдущем этапе. И тут есть два подхода: либо пересчитать её заново, либо запомнить. Первый вариант требует больше времени, второй – больше памяти (количество частиц * количество k-векторов * sizeof(float2)). Я остановился на втором варианте:
__global__ void recip_ewald(int atPerBlock, int atPerThread, cudaMD* md)
// calculate reciprocal part of Ewald summ
// the first part : summ (qiexp(kr)) evaluation
{
int i; // for atom loop
int ik; // index of k-vector
int l, m, n;
int mmin = 0;
int nmin = 1;
float tmp, ch;
float rkx, rky, rkz, rk2; // component of rk-vectors
int nkx = md->nk.x;
int nky = md->nk.y;
int nkz = md->nk.z;
// arrays for keeping iexp(k*r) Re and Im part
float2 el[2];
float2 em[NKVEC_MX];
float2 en[NKVEC_MX];
float2 sums[NTOTKVEC]; // summ (q iexp (k*r)) for each k
extern __shared__ float2 sh_sums[]; // the same in shared memory
float2 lm; // temp var for keeping el*em
float2 ck; // temp var for keeping q * el * em * en (q iexp (kr))
// invert length of box cell
float ra = md->revLeng.x;
float rb = md->revLeng.y;
float rc = md->revLeng.z;
if (threadIdx.x == 0)
for (i = 0; i < md->nKvec; i++)
sh_sums[i] = make_float2(0.0f, 0.0f);
__syncthreads();
for (i = 0; i < md->nKvec; i++)
sums[i] = make_float2(0.0f, 0.0f);
int id0 = blockIdx.x * atPerBlock + threadIdx.x * atPerThread;
int N = min(id0 + atPerThread, md->nAt);
ik = 0;
for (i = id0; i < N; i++)
{
// save charge
ch = md->specs[md->types[i]].charge;
el[0] = make_float2(1.0f, 0.0f); // .x - real part (or cos) .y - imagine part (or sin)
em[0] = make_float2(1.0f, 0.0f);
en[0] = make_float2(1.0f, 0.0f);
// iexp (ikr)
sincos(d_2pi * md->xyz[i].x * ra, &(el[1].y), &(el[1].x));
sincos(d_2pi * md->xyz[i].y * rb, &(em[1].y), &(em[1].x));
sincos(d_2pi * md->xyz[i].z * rc, &(en[1].y), &(en[1].x));
// fil exp(iky) array by complex multiplication
for (l = 2; l < nky; l++)
{
em[l].x = em[l - 1].x * em[1].x - em[l - 1].y * em[1].y;
em[l].y = em[l - 1].y * em[1].x + em[l - 1].x * em[1].y;
}
// fil exp(ikz) array by complex multiplication
for (l = 2; l < nkz; l++)
{
en[l].x = en[l - 1].x * en[1].x - en[l - 1].y * en[1].y;
en[l].y = en[l - 1].y * en[1].x + en[l - 1].x * en[1].y;
}
// MAIN LOOP OVER K-VECTORS:
for (l = 0; l < nkx; l++)
{
rkx = l * d_2pi * ra;
// move exp(ikx[l]) to ikx[0] for memory saving (ikx[i>1] are not used)
if (l == 1)
el[0] = el[1];
else if (l > 1)
{
// exp(ikx[0]) = exp(ikx[0]) * exp(ikx[1])
tmp = el[0].x;
el[0].x = tmp * el[1].x - el[0].y * el[1].y;
el[0].y = el[0].y * el[1].x + tmp * el[1].y;
}
//ky - loop:
for (m = mmin; m < nky; m++)
{
rky = m * d_2pi * rb;
//set temporary variable lm = e^ikx * e^iky
if (m >= 0)
{
lm.x = el[0].x * em[m].x - el[0].y * em[m].y;
lm.y = el[0].y * em[m].x + em[m].y * el[0].x;
}
else // for negative ky give complex adjustment to positive ky:
{
lm.x = el[0].x * em[-m].x + el[0].y * em[-m].y;
lm.y = el[0].y * em[-m].x - em[-m].x * el[0].x;
}
//kz - loop:
for (n = nmin; n < nkz; n++)
{
rkz = n * d_2pi * rc;
rk2 = rkx * rkx + rky * rky + rkz * rkz;
if (rk2 < md->rKcut2) // cutoff
{
// calculate summ[q iexp(kr)] (local part)
if (n >= 0)
{
ck.x = ch * (lm.x * en[n].x - lm.y * en[n].y);
ck.y = ch * (lm.y * en[n].x + lm.x * en[n].y);
}
else // for negative kz give complex adjustment to positive kz:
{
ck.x = ch * (lm.x * en[-n].x + lm.y * en[-n].y);
ck.y = ch * (lm.y * en[-n].x - lm.x * en[-n].y);
}
sums[ik].x += ck.x;
sums[ik].y += ck.y;
// save qiexp(kr) for each k for each atom:
md->qiexp[i][ik] = ck;
ik++;
}
} // end n-loop (over kz-vectors)
nmin = 1 - nkz;
} // end m-loop (over ky-vectors)
mmin = 1 - nky;
} // end l-loop (over kx-vectors)
} // end loop by atoms
// save sum into shared memory
for (i = 0; i < md->nKvec; i++)
{
atomicAdd(&(sh_sums[i].x), sums[i].x);
atomicAdd(&(sh_sums[i].y), sums[i].y);
}
__syncthreads();
//...and to global
int step = ceil((double)md->nKvec / (double)blockDim.x);
id0 = threadIdx.x * step;
N = min(id0 + step, md->nKvec);
for (i = id0; i < N; i++)
{
atomicAdd(&(md->qDens[i].x), sh_sums[i].x);
atomicAdd(&(md->qDens[i].y), sh_sums[i].y);
}
}
// end 'ewald_rec' function
__global__ void ewald_force(int atPerBlock, int atPerThread, cudaMD* md)
// calculate reciprocal part of Ewald summ
// the second part : enegy and forces
{
int i; // for atom loop
int ik; // index of k-vector
float tmp;
// accumulator for force components
float3 force;
// constant factors for energy and force
float eScale = md->ewEscale;
float fScale = md->ewFscale;
int id0 = blockIdx.x * atPerBlock + threadIdx.x * atPerThread;
int N = min(id0 + atPerThread, md->nAt);
for (i = id0; i < N; i++)
{
force = make_float3(0.0f, 0.0f, 0.0f);
// summ by k-vectors
for (ik = 0; ik < md->nKvec; ik++)
{
tmp = fScale * md->exprk2[ik] * (md->qiexp[i][ik].y * md->qDens[ik].x - md->qiexp[i][ik].x * md->qDens[ik].y);
force.x += tmp * md->rk[ik].x;
force.y += tmp * md->rk[ik].y;
force.z += tmp * md->rk[ik].z;
}
md->frs[i].x += force.x;
md->frs[i].y += force.y;
md->frs[i].z += force.z;
} // end loop by atoms
// one block calculate energy
if (blockIdx.x == 0)
if (threadIdx.x == 0)
{
for (ik = 0; ik < md->nKvec; ik++)
md->engCoul2 += eScale * md->exprk2[ik] * (md->qDens[ik].x * md->qDens[ik].x + md->qDens[ik].y * md->qDens[ik].y);
}
}
// end 'ewald_force' function
Надеюсь, вы мне простите, что я оставил комментарии на английском, код практически повторяет последовательную версию. Код даже стал читабельнее за счет того, что массивы потеряли одно измерение: elc/s[i][l], emc/s[i][m] и enc/s[i][n] превратились в одномерные el, em и en, массивы lmc/s и ckc/s – в переменные lm и ck (пропала мерность по частицам, поскольку отпала необходимость хранить это для каждой частицы, промежуточный результат накапливается в shared memory). К сожалению, тут же возникла и проблема: массивы em и en пришлось задать статическими, чтобы не использовать глобальную память и не выделять память динамически каждый раз. Количество элементов в них определяется директивой NKVEC_MX (максимальное количество k-векторов по одному измерению) на этапе компиляции, а runtime используются только первые nky/z элементов. Кроме того, появился сквозной индекс по всем k-векторам и аналогичная директива, ограничивающая общее количество этих векторов NTOTKVEC. Проблема возникнет, если пользователю понадобится больше k-векторов, чем определено директивами. Для вычисления энергии предусмотрен блок с нулевым индексом, поскольку неважно какой именно блок выполнит этот расчет и в каком порядке. Тут может быть надо было использовать готовые функции FFT, раз уж метод основан на нём, но я так и не сообразил, как это сделать.
Ну а теперь попробуем что-нибудь посчитать, да тот же содиум хлорайд. Возьмём 2 тысячи ионов натрия и столько же хлора. Заряды зададим целыми, а парные потенциалы возьмём, например, из этой работы. Стартовую конфигурацию зададим случайно и слегка перемешаем её, рисунок 2а. Объём системы выберем так, чтобы он соответствовал плотности поваренной соли при комнатной температуре (2,165 г/см3). Запустим все это на небольшое время (10’000 шагов по 5 фемтосекунд) с наивным учетом электростатики по закону Кулона и используя суммирование по Эвальду. Результирующие конфигурации приведены на рисунках 2б и 2в, соответственно. Вроде бы в случае с Эвальдом система чуть больше упорядочилась чем без него. Важно также, что флуктуации полной энергии с применением суммирования существенно уменьшились.

Рисунок 2. Начальная конфигурация системы NaCl (a) и после 10’000 шагов интегрирования: наивным способом (б) и со схемой Эвальда (в).
Вместо заключения
Замечу, что структура, получаемая на рисунке, не соответствует кристаллической решетки NaCl, а скорее – решетке ZnS, но это уже претензия к парным потенциалам. Учет же электростатики очень важен для молекулярно-динамического моделирования. Считается, что именно электростатическое взаимодействие ответственно за образование кристаллических решёток, поскольку действует на больших расстояниях. Правда с этой позиции сложно объяснить как при охлаждении кристаллизуются такие вещества как аргон.
Кроме упомянутого метода Эвальда, есть ещё и другие способы учета электростатики, смотрите, например, этот обзор.
Похожие статьи
 Физики впервые создали квантовый материал, работающий при комнатной температуре
Физики впервые создали квантовый материал, работающий при комнатной температуре Забытый шедевр среди операционных систем
Забытый шедевр среди операционных систем Как я выпустила альбом электроники, созданный на основе звуков рек?
Как я выпустила альбом электроники, созданный на основе звуков рек? Как работают электрические рыбы?
Как работают электрические рыбы? От зефира к фаллосу: как Ридли Скотт спас «Чужого»
От зефира к фаллосу: как Ридли Скотт спас «Чужого» В США разрешили развертывание первой орбитальной группировки космических отражателей
В США разрешили развертывание первой орбитальной группировки космических отражателей Уход Солнца
Уход Солнца WebTV: как интернет пришел на экраны телевизоров
WebTV: как интернет пришел на экраны телевизоров