Главный приз — Полный геном.

Ранее мы уже делились полезной информацией и ссылками, которые могут пригодиться для работы с биоинформатическими данными. Советуем сначала прочитать предыдущие статьи, если вы их пропустили.
Дисклеймер
Работа с генетическими данными проводится на Unix системах (Linux, macOS), так как некоторые команды и ПО недоступны на Windows. Поэтому для пользователей Windows одним из самых простых решений будет арендовать виртуальную машину с Linux.
Все описанные ниже операции выполняются в командной строке — терминале. Перед началом выполнения узнайте, как работать в терминале под управлением вашей ОС и использовать команды, так как некоторые из них могут потенциально навредить ОС и вашим данным.
Необходимое ПО
Мы собрали образ виртуальной машины (ВМ) со всем необходимым ПО на Яндекс.Облаке. Инструкции по настройке ВМ и установке ПО можно найти в предыдущей статье с Задачей № 1.
На этот раз вам потребуется построить двумерную точечную диаграмму, используя данные, полученные методом анализа главных компонент. Предлагаем построить эту диаграмму с помощью любого удобного для вас ПО: Excel, Google Sheets, Python, R и прочие.
Для выполнения задачи понадобится программный пакет Plink 1.9. Если вы его еще не установили (и не выполняли Задачу № 1), прочитайте предыдущую статью. В ней содержатся инструкции по установке. Для участия в новогоднем конкурсе 2019 необходимо выполнение всех задач!
Взять на заметку
Анализ главных компонент (PCA, principal component analysis) — это один из алгоритмов машинного обучения без учителя, когда машина самостоятельно ищет паттерны в данных. В генетике PCA позволяет кластеризовать образцы по данным генотипирования в N-мерном пространстве (обычно двумерном), где получаемые главные компоненты наиболее точно объясняют вариабельность генетических данных от образца к образцу.
При проведении такого анализа образцы одной популяции обычно образуют кластер, размер и гладкость границ которого зависят от схожести образцов внутри данной популяции. Образцы из разных популяций алгоритм скорее всего определит в разные кластеры. А образцы из близких популяций, которые относятся к одной суперпопуляции, например, EAS — East Asian, как на Рисунке 1, будут определены в близкие друг к другу или даже в пересекающиеся кластеры.

Рисунок 1. Родословная используемых в VCF образцов (квадрат соответствует мужскому полу, круг — женскому). Пунктирная линия соответствует неустановленному родству второго порядка.
Подобный анализ используется для определения популяционной принадлежности по данным генотипирования. Для этого необходим референсный датасет, который состоит из образцов с уже известным происхождением. Вывод о популяционной принадлежности можно сделать по тому, к какому кластеру из известных образцов ближе всего изучаемые данные.
Если упрощать, то суть PCA анализа заключается в том, что известны попарные расстояния между точками в многомерном пространстве, и надо расположить эти точки в пространстве меньшей размерности так, чтобы новые попарные расстояния минимально отличались от изначальных. Снижение размерности упрощает анализ данных, но чем сильнее мы ее снижаем, тем сильнее новые расстояния между точками отличаются от изначальных. Поэтому задача PCA анализа включает также поиск компромисса между точностью и простотой анализа. Все как в жизни.
В основе простейшего варианта выполнения PCA на генетических данных лежит идентичность некоторых аллелей, которая может быть разделена на два подтипа: IBS (identity by state) и IBD (identity by descent). IBS означает идентичность определенных аллелей у двух людей, но не обязательно подразумевает факт какого-либо родства между ними. IBD, напротив, говорит об идентичности аллелей из-за наличия общего предка и, соответственно, родства.
IBD аллели однозначно являются IBS аллелями, в то время как обратное утверждение неверно. Однако, нужно учитывать, что в какой-то момент времени мы произошли от общего предка, поэтому некоторые аллели могут являться IBD. В PCA анализе ниже используется только концепция IBS, хотя в более сложных анализах в ней учитываются тесты статистической значимости, фенотипические ограничения, размеры кластеров, возраст и пол человека, а также дополнительная информация о популяционной структуре.
Чем больше у двух образцов количество отличающихся аллелей, тем меньше они похожи и тем дальше находятся друг от друга. Значение величины IBS для таких образцов будет низким. А вот для родителей и их детей величина IBS будет очень высокой.
Зная значения IBS для каждой пары образов в датасете, можно провести PCA анализ, чтобы посмотреть, как они кластеризуются.
В генетическом тесте «Атлас» используется намного более сложный алгоритм определения популяционной представленности в данных генотипирования.
Используемые данные
Напоминаем, что в руководстве используются специально отобранные нами открытые данные из проекта «1000 Геномов». Для анализа были выбраны 10 образцов с информацией о генотипах ~85 миллионов вариаций, которые получены путем анализа данных NGS с выравниванием на версию генома GRCh37. Родственные связи и популяции данных образцов указаны на Рисунке 1.
Построение популяционных кластеров
Используйте полученные ранее в Задаче № 1 три файла в формате Plink:
CEI.1kg.2019.demo.subset.bed
CEI.1kg.2019.demo.subset.bim
CEI.1kg.2019.demo.subset.famОпределите попарное расстояние между всеми 10 образцами в обучающем датасете и проведите PCA на основе IBS (identity by state). Это можно сделать следующим образом:
# Перейдите в нужную директорию
plink —bfile CEI.1kg.2019.demo.subset —genome —out CEI.1kg.2019.demo.subset.similarities
plink —bfile CEI.1kg.2019.demo.subset —read-genome CEI.1kg.2019.demo.subset.similarities.genome —cluster —mds-plot 10 —out CEI.1kg.2019.demo.subset.clustering
Параметр —genome как раз отвечает за попарный подсчет IBS/IBD между всеми образцами в датасете. Параметр —read-genome — это полученная ранее матрица попарных расстояний, а параметры —cluster —mds-plot 10 отвечают за PCA анализ и вывод его результатов в таблицу по 10 первым главным компонентам. По сути, это координаты каждого образца в 10-мерном пространстве.
Последняя команда создаст в папке 4 файла:
CEI.1kg.2019.demo.subset.clustering.cluster1
CEI.1kg.2019.demo.subset.clustering.cluster2
CEI.1kg.2019.demo.subset.clustering.cluster3
CEI.1kg.2019.demo.subset.clustering.mdsНам будут необходимы два последних файла из списка.
На Рисунке 2 показано, как выглядит полученный на обучающем датасете MDS файл. Поля FID (Family ID) и IID (Individual ID) соответствуют идентификаторам семьи и отдельного образца. Поля С1 — С10 содержат значения каждой из десяти главных компонент для каждого образца, где компонента С1 максимально объясняет вариабельность генетических данных анализируемых образцов, а С10 — минимально.

Рисунок 2. MDS файл со значениями 10 главных компонент для каждого образца.
При построении точечной диаграммы с использованием двух компонент (в двумерном пространстве) можно обнаружить кластеры, соответствующие популяционной принадлежности образца. На Рисунке 3 приведены точечные диаграммы для пар главных компонент С1xС2, С2xС3 и С1xС3. При сравнении полученных кластеров с референсной популяционной принадлежностью (Рисунок 1) наиболее высокую точность (100%) показывает пара первых двух компонент С1—С2, верно разделяющая все образцы в соответствии с их заявленной в проекте «1000 Геномов» популяционной принадлежностью. Однако всегда имеет смысл сравнивать полученные результаты для нескольких пар компонент из-за возможного перекрытия или разделения реальных кластеров.

Рисунок 3. Точечные диаграммы расположения образцов по парам главных компонент; расположение маркеров было незначительно изменено для предотвращения их перекрывания друг другом.
Построение 3D диаграмм с использованием первых трех главных компонент также может помочь в определении кластеризации, но не всегда. Например, построение такой диаграммы для данных на Рисунке 3 позволяет выделить 4 кластера, в которых образцы из популяций PUR и KHV кластеризуются в соответствии с популяционной принадлежностью, а образцы из популяции CDX разделяются на два кластера(Рисунок 4). Это заметно и на Рисунке 3 в координатах С2хС3 и С1хС3.
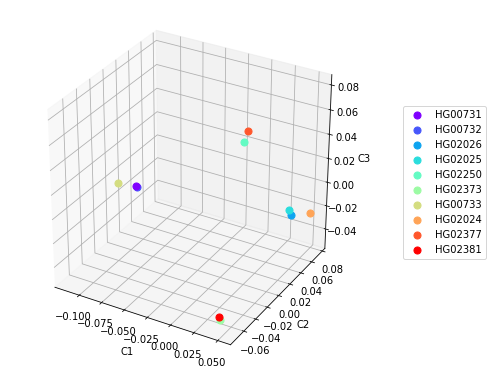
Рисунок 4. Точечные диаграммы расположения образцов по трем главным компонентам.
Подобные противоречивые результаты анализа могут объясняться небольшим количеством образцов, так как значения главных компонент каждого образца отличны для разных по размеру и составу датасетов и при включении дополнительных образцов из разных популяций результат кластеризации может измениться. Также потенциально возможно наличие ошибок при создании датасета и предоставлении референсных данных о популяционной принадлежности образцов, однако в проекте «1000 Геномов» вероятность такой ситуации достаточно низкая.
MDS файл не использует табуляцию или запятые в качестве разделителей, поэтому для удобства поправьте его формат. Используйте tab или csv в качестве второго аргумента:
# Перейдите в нужную директорию
convert_plink_delimiter.sh CEI.1kg.2019.demo.subset.clustering.mds tab
Команда создаст файл CEI.1kg.2019.demo.subset.clustering.mds.tab, который можно скачать и построить точечные диаграммы, подобные представленным на Рисунке 3. Сравните полученные результаты, они должны быть идентичны указанным выше.
Построение кластеризационного дерева
Дополнительно оценить кластеризацию образцов можно с помощью бинарного дерева, представляющего кластеризационную информацию об образцах в дискретном виде. Информация об этом дереве содержится в файле CEI.1kg.2019.demo.subset.clustering.cluster3 (Рисунок 5).
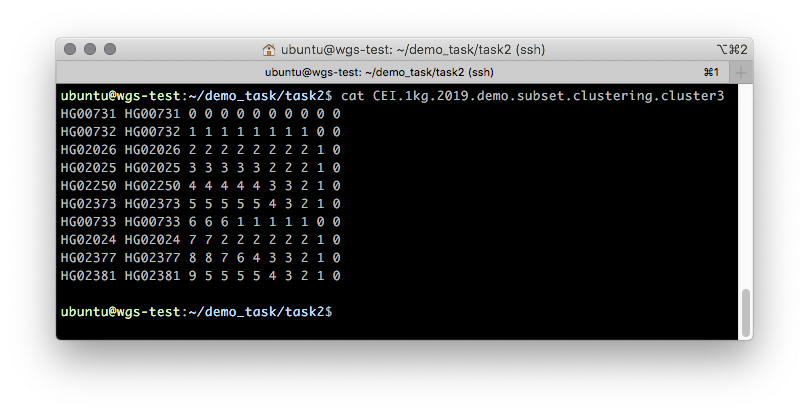
Рисунок 5. Примерное содержимое .cluster3 файла, описывающего процесс пошаговой кластеризации образцов от 1 кластера до N, где N — количество образцов.
Первые две колонки данного файла содержат FID и IID. Кластерную принадлежность описывают все остальные. Читать данный файл следует справа налево по колонкам с шагом в одну колонку: изначально все образцы принадлежат одному кластеру “0” — самая правая колонка. При разбиении на два кластера (на втором шаге, во второй колонке) появляется два кластера: “0” и “1”, где кластеру “0” принадлежат образцы HG00731, HG00732 и HG00733, а кластеру “1” — все остальные. Иллюстрация такого разбиения показана на Рисунке 6.
Из дерева можно сделать вывод о популяционной принадлежности образцов (Рисунок 1). Помимо этого, построение данного дерева позволяет установить близость отдельно взятых популяций, а именно вхождение популяции CDX и KHV в одну суперпопуляцию EAS (уже на первом шаге разбиения суперпопуляции EAS и AMR обособлены в двух существующих ветвях). Также построение кластеризационного дерева может помочь скорректировать неоднозначные результаты визуализации образцов по главным компонентам.
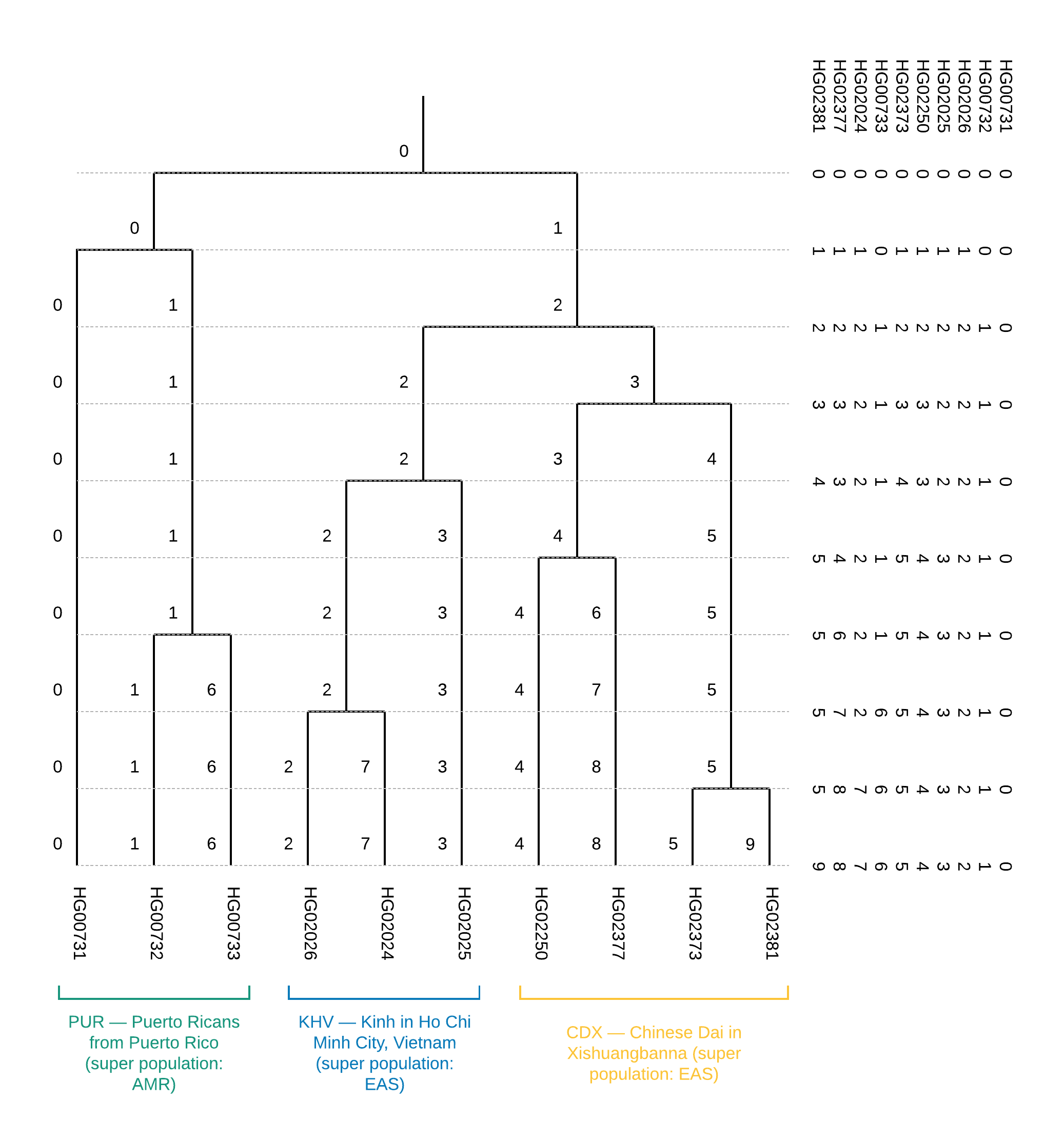
Рисунок 6. Бинарное дерево кластеризации для обучающего датасета из 10 образцов: справа приведено содержимое файла CEI.1kg.2019.demo.subset.clustering.cluster3 (справа налево в файле тождественно сверху вниз на рисунке).
Второе задание конкурса
Используйте тестовый датасет из 12 образцов Data/Test/CEI.1kg.2019.test.vcf.gz и пример выше (Рисунок 5), чтобы построить бинарное дерево кластеризации по полученному вами .cluster3 файлу и приложить его к решению. Проанализируйте полученное дерево и сделайте выводы о количестве суперпопуляций, представленных в тестовом датасете.
Определите популяционную кластеризацию 12 образцов из тестового датасета по анализу главных компонент C1, С2 и С3 с учетом построенного дерева и укажите это на построенной в Задаче № 1 родословной, ограничивая отдельные популяционные блоки (по аналогии с Рисунком 1). Образцы, которые не показали наличие родственных связей в Задаче № 1, необходимо таким же образом расположить внутри полученных блоков на схеме, не соединяя их линиями с другими образцами. Не забудьте приложить построенные вами точечные диаграммы.
Ответы присылайте на почту wgs@atlas.ru до 26 декабря до 23:59. Еще одна задача будет скоро опубликована, а итоговые результаты по задачам появятся 28 декабря. Победитель получит тест Полный геном, а второе и третье места — генетический тест «Атлас». Также будут специальные призы от Яндекс.Облако. Бывшие и настоящие сотрудники Атласа в конкурсе не участвуют 😉
")

