Проект aztotmd был создан для реализации собственных идей относительно компьютерного моделирования. Основан на классической молекулярной динамике и содержит основной функционал для классических расчётов, но также и ряд экспериментальных особенностей: непостоянное поле сил и излучательный термостат. Программа распараллелена с помощью технологии CUDA, на хорошей десктопной видеокарте можно выполнить прогон в миллион шагов для нескольких тысяч атомов часов за 5-6. Часть алгоритмов была описана ранее: основа, метод Эвальда и внутримолекулярное взаимодействие. Сейчас представляю инструкцию по работе с программой.
1 Возможности и ограничения
На данный момент поддерживается только параллельная CUDA-версия, поэтому соответствующие ограничения на железо – видеокарта от NVidia с computational capability > 2.2. Интегрирование уравнений движения скоростным алгоритмом Верле (Velocity Verlet). Опции:
-
(на данный момент) только периодические граничные условия и только в форме прямоугольного параллелепипеда;
-
парные потенциалы: 6 обычных (включая Леннарда-Джонса, Букингема, Борна-Майера-Хаггинса и др.) и 1 температуро-зависимый для излучательного термостата. Если не боитесь кодить, можно легко добавить свои (поддерживается до 5 параметров). Отдельный cutoff для каждого потенциала;
-
3 способа учета электростатики: наивный, суммирование по Эвальду (Particle mesh Ewald, PME) и метод Феннеля и Гецельтера;
-
2 термостата: Нозе-Гувера и излучательный;
-
валентные связи (5 потенциалов, возможность добавления своих);
-
валентны углы (пока только в форме гармонического косинуса, легко добавить свои);
-
внешнее электрическое поле с постоянным градиентом;
-
«замороженные» типы атомов;
-
возможность динамического образования/удаления валентных связей (включая водородные) и валентных углов;
-
электронный перенос между атомами;
-
вывод статистики (различные энергии, кинетическая температура, давление и т.д.), радиальных функций распределения (опционально можно выводить не только конечную, но и через определённый шаг), траекторий движения (опционально – с расширенной информацией), конечной конфигурации (+ связей и углов), статистики по связям.
2 Установка
Самый простой вариант – если у вас Windows (проверял только на 10-м) – можно скачать экзешник отсюда.
Если у вас установлена MS Visual Studio и с ней интегрирована NVidia CUDA можете попробовать самостоятельно скомпилировать проект, скачав репозиторий на гитхабе, используйте ветку develop, там последняя версия. Открывайте проект aztotmd, не aztotmd_serial. У меня 11 версия CUDA, если у вас другая – возможно потребуется отредактировать файл проекта вручную в текстовом редакторе, заменив соответствующие цифры.
Наконец, если у вас не Windows, можете попробовать написать makefile, собирающий все модули, и скомпилировать проект (главный файл main.cu, не main.cpp). Скорее всего, придётся подредактировать и сам код, хотя я старался свести использование сторонних библиотек к минимуму.
Изначально я пытался подружить nvcc-компилятор со средой CodeBlocks, но у меня ничего не вышло, пришлось устанавливать MS Visual Studio. Если вам удалось встроить кудовский компилятор в какую-нибудь среду, отличную от студии – напишите.
3 Запуск
Программа не имеет графического интерфейса, ввод и вывод информации осуществляется через текстовые файлы. Для работы программы необходимо, чтобы в директории с экзешником (исполняемым файлом) находились входные файлы (4 из которых — обязательны):
-
atoms.xyz (обязательный) – размер бокса и координаты частиц (конфигурация системы);
-
field.txt (обязательный) – характеристики частиц, поле сил;
-
control.txt (обязательный) – общие директивы;
-
cuda.txt (обязательный) – директивы, касающиеся видеокарты;
-
bonds.txt (опциональный) – список валентных связей;
-
angles.txt (опциональный) – список валентных углов.
3.1 Файл atoms.xyz. Текстовый файл в соответствии с форматом XYZ. Первая строка – число частиц. Вторая строка в оригинальном формате зарезервирована под комментарий, но для aztotmd там должны находится характеристики бокса: тип бокса (на данный момент поддерживается только орторомбический периодический бокс, символ 1) и его размеры по осям x, y и z (в ангстремах, Å).
Последующие строки – тип и координаты частиц (x y z) в ангстремах (Å). Все типы частиц должны быть определены в файле field.txt. Разделитель – любое количество пробельных символов (включая табуляцию или перенос строки). Пример файла atoms.xyz (показаны только несколько первых строк):
3361
1 36.500000 36.500000 36.500000
V 2.935293 1.855377 0.381776
Od 2.663695 2.704217 1.708393
V 2.423310 3.419446 3.390218
Od 1.036296 1.359655 0.216446
Oc 3.856887 0.492592 1.200182Формат *.xyz поддерживается многими молекулярными редакторами. Мне очень нравится программа Diamond, весь функционал, кроме сохранения во внутреннем формате поддерживается бесплатной версией. Сгенерировать файл с начальной конфигурацией можно также с помощью нашего сайта, поддерживаются геометрии кристаллических решёток NaCl (чередование атомов в шахматном порядке по 3-ём осям) и ZnS (каждый атом окружён 4-мя соседями по вершинам тетраэдра). Если периодические системы не нужны, можно по-прежнему воспользоваться данным генератором, просто взять произвольные потенциалы и дать небольшой прогон при высокой температуре, атомы перемешаются.
3.2 Файл field.txt. Текстовый файл, содержащий поле сил: все взаимодействия, характеристики частиц и т.п. Состоит из разделов и директив. Разделы начинаются с соответствующего ключевого слова и числа, указывающего количество элементов раздела. Разделы:
3.2.1 Раздел spec – единственный обязательный раздел, где приводятся характеристики всех используемых в моделировании типов частиц. Укажите ключевое слово spec и число типов частиц. Далее построчно перечисляются характеристики каждого типа частиц: название (произвольное, до 7 непробельных символов), соответствующий «остов» (произвольное, до 7 непробельных символов, может совпадать с названием типа частицы), массу (в а.е.м.), заряд (в зарядах протона), «собственную энергию» (в эВ, нужна в очень редких случаях, можно указать 0). Указание остова для каждого типа частиц было введено, чтобы можно было группировать сходные типы частиц и выводить обобщенную статистику по ним (в первую очередь, радиальные функции распределения). Например, в системе есть два типа ионов железа: Fe2+ и Fe3+, каждый со своими парными потенциалами, но радиальная функция распределения интересует обобщенная, тогда можно отнести эти ионы к одному остову. Пример раздела spec:
spec 5
O2- O 16.0 -2.0 0.0
Od O 16.0 -1.2 0.0
P5 P 31.0 5.0 0.0
P3 P 31.0 4.2 0.0
V5 V 51.0 5.0 0.03.2.2 Раздел frozensp заставляет частицы определенного типа не двигаться. Укажите ключевое слово frozensp и число «замороженных» типов частиц, после чего перечислите их. Пример раздела:
frozensp 2
La Sc3.2.3 Раздел vdw – межмолекулярные парные потенциалы. Построчно вводите характеристики каждого парного потенциала: типы частиц, между которыми действует данный парный потенциал; ключевое слово, обозначающее функциональную форму парного потенциала; радиус обрезания (в Å); параметры парного потенциалов. Поддерживаемые формы парных потенциалов и, соответствующее им, количество параметров указаны в таблице 1. Все значения указываются в Å, эВ и производных от них единицах. Все типы частиц должны быть определены в разделе spec. Пример раздела vdw:
vdw 3
V3 Ot elin 6.0 80.0 0.3 0.75
V3 Od bmhs 2.0 2.68839 3.27417 1.78352 18.01372 -1.06927
V4 Od buck 2.5 1290.56 0.34039 0.0Парные потенциалы (ключевые слова): Леннарда-Джонса (lnjs), Букингема (buck), Борн-Майера-Хаггинса (bmhs), для щелочных галлидов (p746), отталкивающая экспонента + линейная функция (elin), отталкивающая экспонента + обратная функция (einv), температурно-зависимый потенциал (surk). Последний потенциал (surk) для своей работы требует излучательный термостат и задание радиуса частиц, как функции от «термальной» энергии (см. раздел 6.2 Излучательный термостат).
Таблица 1. Поддерживаемые межмолекулярные парные потенциалы.
|
ключевое слово |
потенциал |
порядок перечисления параметров |
|
lnjs |
U = 4ε[(σ/r)12 — (σ/r)6] |
ε, σ |
|
buck |
U = A exp (-r/ρ) – C/r6 |
A, ρ, C |
|
bmhs |
U = A exp[B(σ – r)] – C/r6 – D/r8 |
A, B, σ, C, D |
|
p746 |
U = A/r7 – B/r4 – C/r6 |
A, B, C |
|
elin |
U = A exp (-r/ρ) – Cr |
A, ρ, C |
|
einv |
U = A exp (-r/ρ) – C/r |
A, ρ, C |
|
surk |
U = rirj[C1(rirj)2/r7 – C2/(ari + brj)/r6] |
C1, C2, a, b |
3.2.4 Раздел red-ox. Предназначен включения окислительно-восстановительных процессов (переноса электрона) в молекулярную динамику. НЕ ЯВЛЯЕТСЯ классической молекулярной динамикой. Укажите ключевое слово red-ox и число «окислительно-восстановительных последовательностей». Далее построчно введите последовательности: число типов частиц в последовательности с их перечислением, начиная от наивысшей степени окисления по порядку. Степени окисления каждой следующей частицы в последовательности должны различаться на 1, иначе разбейте последовательность на несколько. Само наличие этого раздела не активирует электронный перенос, для этого необходимо использовать директиву ejump в файле control.txt (см. раздел 3.3). Пример раздела red-ox:
red-ox 4
4 V5 V4 V3 V2
2 Cl0 Cl-
2 Mn7 Mn6
3 Mn4 Mn3 Mn23.2.5 Раздел bonds – описание типов внутримолекулярных (валентных) связей. Перечислите все возможные типы валентных связей, которые есть (или могут быть) в вашей системе. Построчно запишите свойства каждого типа связи. Строка, описывающая потенциал валентной связи, содержит:
-
порядковый номер (нумеруйте типы связей по порядку, начиная с 1*);
-
типы частиц, между которыми действует связь;
-
ключевое слово, обозначающее функциональную форму потенциала (таблица 2);
-
параметры потенциала, таблица 2;
-
ключевое слово, обозначающее эффект при сокращении длины связи: con или mut**:
-
con – связь не меняется,
-
mut – связь меняется при сокращении, в этом случае далее нужно указать минимальную длину связи (в Å), ниже которого связь меняет свой тип и номер нового типа связи;
-
-
ключевое слово, обозначающее эффект при растяжении связи: con, br или mut**:
-
con – связь не меняется;
-
br – связь обрывается при растяжении, после br укажите максимальную длину связи (в Å) и типы частиц, в которые превращаются частицы, образующие связь (в порядке, соответствующем начальным типам);
-
mut – связь превращается в другую при растяжении, укажите максимальную длину связи и номер нового типа связи.
-
Примечания:
* нумерация нужна для удобства восприятия. Нумеруйте связи по порядку, начиная с 1, поскольку программа всё равно их так пронумеруют, какие бы вы числа не указывали.
** в классической молекулярной динамике связи не рвутся и не меняются, если вы хотите оставаться в рамках классической МД в обоих случаях укажите ключевое слово con.
Таблица 2. Поддерживаемые потенциалы валентных связей.
![aztotmd: молекулярная динамика [+ непостоянное поле сил] [+ излучательный термостат]. CUDA-версия. Руководство](https://habrastorage.org/getpro/habr/upload_files/dec/042/4ca/dec0424ca4955cb841c01d8e40045fcc.svg "aztotmd: молекулярная динамика [+ непостоянное поле сил] [+ излучательный термостат]. CUDA-версия. Руководство")
![U=D[1-exp(-alpha(r-r_0))]^2-C](https://habrastorage.org/getpro/habr/upload_files/b17/c03/2bc/b17c032bcb9c14838780da8f5edf9c55.svg)
![U=D[1-exp(-alpha(r-r_0))]^2-C-frac E{r^{12}}](https://habrastorage.org/getpro/habr/upload_files/c82/500/8fb/c825008fbbf42ddb1ecb8bb34fba6007.svg)
Пример раздела bonds:
bonds 4
1 V3 Od harm 50.0 1.62 con con
2 V Od mors 10.0 1.4959 1.584 10.0 con mut 1.8 3
3 V+ Ot mors 6.496 1.4959 1.791 6.496 mut 1.8 2 con
4 Hh Ot mors 0.22 1.0 2.04 0.22 con br 2.2 H Ot3.2.6 Раздел h-bonds. В некоторых случаях часть связей из раздела bonds необходимо пометить как «водородные». Это связано со спецификой алгоритмов образования и разрушения связей. Если у вас нет образования или разрушения связей, этот раздел не нужен. В заголовке раздела укажите число водородных связей, затем перечислите пары номер связи и тип атома, считающийся атомов водорода. Пример раздела h-bonds:
h-bonds 4
30 H+h 31 H+h 33 H2h 34 H2hВодородные связи обрабатываются несколько по-другому, чем обычные. При их создании не образуются валентные углы. Подробнее см. раздел 6.1 Связывание.
3.2.7 Раздел evol_bonds. Этот раздел также нужен в очень редких случаях. При изменении типов частиц программа автоматически меняет тип связи между частицами (если связь была). В том случае, когда есть несколько возможных вариантов связей между частицами, этот раздел позволяет указать, как конкретно изменится тип связи. Укажите ключевое слово evol_bonds и количество связей, для которых вы хотите определить новый тип связи при изменении. Далее укажите через — (знак минуса) пары типов связей старый-новый, например:
evol_bonds 2
35-39 29-36Эта запись означает, что если частицы, связанные связью типа 35, изменили свой тип, то Программа в первую очередь попробует изменить тип связи на 39. Важно! Программа может изменить тип связи на указанный, только если он соответствует новым типам частиц. Подробнее см. раздел 6.1 Связывание.
3.2.8 Раздел linkage. Для того, чтобы частицы самопроизвольно образовывали связи между собой, объявите раздел linkage. Напишите ключевое слово linkage и количество пар частиц, способных образовать связь. Затем построчно укажите пару частиц, минимальное расстояние связывания и тип образующейся связи. Типы частиц и типы связей должны быть определены в секциях spec и bonds, соответственно:
bonds 1
1 V3 Od harm 50.0 1.62 con con
linkage 1
V5 O2- 1.7 1данном примере, частицы V5 и O2- оказавшись на расстоянии 1.7 Å и меньше связываются с образованием связи типа 1 (раздел bonds), при этом частицы превращаются в V3 и Od, соответственно, как указано в объявлении связи. Следите за тем, чтобы порядок типов частиц в секциях bonds и linkage соответствовал. Подробнее см. раздел 6.1 Связывание.
3.2.9 Раздел angles определяет типы валентных углов. Построчно запишите свойства каждого потенциала валентного угла: порядковый номер, центральный тип частицы, ключевое слово парного потенциала (на данный момент поддерживается только hcos – гармонический косинус), параметры потенциала (для гармонического косинуса: константа жесткости в эВ и косинус равновесного угла). Пример секции angles:
angles 1
1 Oc hcos 5.0 -0.333263.2.10 Раздел angle_forming позволяет создавать валентные углы прямо в ходе численного эксперимента. Напишите ключевое слово angle_forming и количество типов частиц, для которых автоматически создаются валентные углы. Далее в каждой строчке указывает тип частицы и тип потенциала валентного угла. Типы частиц и потенциалы валентного должны быть определены в секциях specs и angles:
angle_forming 1
Oc 1Данная запись означает, что как только в системе возникла частица Oc, то будут автоматически созданы валентные углы, с вершиной в этой частице. Остальные атомы в валентных углах те – что связаны валентными связями с Oc. Подробнее см. раздел 6.1 Связывание.
Программа поддерживает также заранее заданные списки валентных связей и углов. Для этого укажите директивы bond_list 1 и angle_list 1, соответственно:
bond_list 1
angle_list 1 в этой случае при старте Программа считает перечень валентных связей и углов из файлов bonds.txt и anlges.txt, соответственно.
3.2.11 Раздел radii. Очередная «экспериментальная» фича, необходимая для реализации собственной концепции температуро-зависимого поля сил в рамках «излучательного термостата». Будем считать, что температура вещества проявляется в энергетическом возбуждении атомов. Величина этой энергии вводится здесь, и будем считать, что эта энергия запасается в виде увеличения Боровского радиуса относительно основного состояния. Для связи радиуса с энергией предложена формула:
r = A / (B — H),
где A и B – константы, H – энергия теплового возбуждения. В основном состоянии H = 0 и радиус равен A/B. Раздел radii позволяет задать константы A и B (и максимальную H). Построчно перечислите частицы, затем параметры A, B и ограничение по H (в эВ):
radii 2
Na 27.3 47.31 0.2
Cl 37.8 47.31 0.23.3 Файл control.txt. Текстовый файл, содержащий общие директивы, порядок перечисления директив неважен. Ниже дан перечень директив (символы N и f означают целое или дробное число, соответственно; разные варианты даны через /, в [] – необязательный параметр):
-
timestep f – (обязательная) задать величину шага интегрирования уравнений движения равной f пикосекунд (пс), обычно 10-4 – 10-3;
-
nstep N – (обязательная) задать число шагов (циклов) МД равным N;
-
nequil N — задать число шагов предварительной релаксации системы равным N;
-
eqfreq N — Масштабировать температуру каждые N шагов в ходе периода релаксации (директива nequil);
-
temperature f1 none / radi / nose f2 – (обязательная) задать температуру равной f1 (в градусах Кельвина). После температуры идёт указание на используемый термостат:
-
none – без термостата, температура игнорируется, NVE-ансамбль;
-
radi – «излучательный термостат», подробнее в разделе 6.2;
-
nose – термостат Нозе-Гувера с константой релаксации f2 пикосекунд;
-
-
init_vel zero / gaus / const f1 f2 f3 / keng f – (обязательная) задать начальное распределение скоростей: zero – задать нулевыми; gaus – согласно распределению Максвелла; const – все частицы обладают одним вектором скорости (f1, f2, f3); keng – модуль скорости для всех частиц соответствует кинетической энергии f эВ, направление случайное;
-
reset_vels N – занулять скорости частиц каждые N шагов;
-
permittivity f — задать диэлектрическую проницаемость равной f, используется для вычисления Кулоновского взаимодействия;
-
elec none / dir f / pme f1 f2 N1 N2 N3 / fenn f1 f2 – (обязательная) способ расчета электростатики:
-
none – без электростатики, заряды частиц игнорируются (кроме как для внешнего поля, см. директиву elecfield);
-
dir f – наивный закон Кулона с радиусом обрезания f Å;
-
pme f1 f2 N1 N2 N3 – использовать суммирование по Эвальду, f1 – радиус обрезания в действительном пространстве, f2 – параметр α, N1 N2 N3 – число k-векторов по осям Ox, Oy и Oz, соответственно;
-
fenn – метод Феннеля и Гезельтера, f1 – радиус обрезания, f2 – параметр α (рекомендую использовать именно эту директиву с радиусом обрезания 8-9 Å и α = 0.4).
-
-
cell_list f – (обязательная) задать размер ячейки в алгоритме cell_list равным f Å (очень важный параметр, влияющий на скорость вычисления. Для конденсированных сред попробуйте порядка 2-4 Å, для разреженных газов, где среднее расстояние между частицами огромное лучше брать десятки ангстрем. Попробуйте разные значения на небольшом количестве шагов и выберите наибыстрейший вариант);
-
rdf f1 f2 N1 N2 [nucl] – (обязательная) директива для расчета радиальной функции распределения (RDF): f1— радиус обрезания (Å), f2 – шаг (Å), сбор статистики каждые N1 шагов, выводить промежуточные результаты каждые N2 шагов. Ключевое слово nucl указывает, что нужно также собирать RDF обобщая атомы по остовам.
-
eJump N f1 min/metr/eq f2 — выполнять процедуру электронного переноса каждые N шагов, на расстоянии до f1 Å (критерием принятия переноса является: min – минимизации энергии; metr – схема Метрополиса; eq – равенство энергии (принцип Франка-Кондона) с допустим отклонением f2 эВ).
-
elecfield f1 f2 f3 — задать постоянный градиент внешнего электрического поля f1, f2 и f3 Вольт/Å вдоль осей Ox, Oy и Oz соответственно. Появляется добавочное слагаемое в потенциальную энергию U = q(f1x + f2y + f3z) и на частицы действует дополнительные силы Fi = -q*fi;
-
stat N – (обязательная) Выводить статистику каждые N шагов. С этой же частотой часть статистики выводится на экран;
-
ncn N – получить распределение координационных чисел по остовам (см. файл nCN.dat) для финальной конфигурации, N – количество пар для вывода. Пары перечисляются в формате <остов> <лиганд>
. Пример:
ncn 3
Fe O 1.6
Mn O 1.7
Fe Fe 3.5-
traj xy N1 N2 N3 N4 – записывать файл с траекториями движения (проекция на Oxy). N1 – с какого шага начинать записывать, N2 – с каким интервалом, N3 – начиная с какого номера атома, N4 – сколько атомов.
3.4 Файл cuda.txt.
Перечисление директив, касающихся видеокарты.
-
multproc N – число используемых мультипроцессоров, установите 0, если собираетесь использовать все имеющиеся. Актуально для интегратора движения, обработки связей и углов. Вычисление межмолекулярных взаимодействий происходит с использованием всех мультипроцессоров.
-
singproc N – число процессоров в составе одного мультипроцессора. Почему-то эту информацию невозможно получить методом GetDeviceProperties, поэтому узнайте её из спецификации своей видеокарты.
-
nthread a N – число потоков на блок в 1ой части алгоритма cell list, рекомендуемое (и дефолтное) значение = 16.
-
nthread b N – число потоков на блок в 2ой части алгоритма cell list, рекомендуемое (и дефолтное) значение = 32.
-
bindtraj threads N1 N2 — количество атомов на поток и количество потоков в блоке для вывода bindtraj (связанных траекторий), рекомендуемые значения 1 и 32, соответственно.
-
nstep stat N – количество итераций статистики, хранимых на видеокарте, до вывода в файл.
-
nstep msdstat N — количество итераций msd-статистики, хранимых на видеокарте, до вывода в файл.
-
nstep bondstat N — количество итераций статистики по связям, хранимых на видеокарте, до вывода в файл
-
nstep traj N — количество итераций траекторий, хранимых на видеокарте, до вывода в файл
-
nstep bindtraj N — количество итераций «связанных траекторий», хранимых на видеокарте, до вывода в файл
3.5 Файл bonds.txt
Текстовый файл, позволяющий задать валентные связи между частицами. Первая строчка – число связей. Далее построчно – индексы двух атомов, связанных связью (нумерация с нуля, согласно файлу atoms.xyz) и тип связи согласно разделу bonds файла field.txt (нумерация с единицы).
3.6 Файл angles.txt
Аналогично файлу bonds.txt позволяет задать валентные углы. Указывается число валентных углов и построчно 4 числа: индексы атомов (первым указывается центральный атом!) и тип валентного угла из раздела angles файла field.txt.
4 Выполнение программы
Программа будет считать, пока не выполнит все итерации, указанные директивой nstep. Можно остановить счет досрочно: Esc — с полноценным завершением программы или Ctrl+C – сбросить. Результат работы программы – набор текстовых файлов.
5 Выходные файлы
5.1 Файлы revcon.xyz, revbonds.txt, revangles.txt – аналоги входных файлов atoms.xyz, bonds.txt и angles.txt, соответственно. Содержат информацию о координатах атома, связях и углах в конце моделирования. Эти файлы можно переименовать в atoms.xyz, bonds.txt и angles.txt и начать следующее моделирование с того состояния (за исключением скоростей и сил), на котором остановились в этом. Остальные файлы – это также текстовые файлы с табуляцией в качестве разделителя. Удобно открывать табличными редакторами, такими как Excel (я предпочитаю Origin), но есть и куча бесплатных аналогов.
5.2 Файл stat.dat – статистика о системе. Первые две колонки: время (в пс) и номер шага. Остальные колонки:
-
кинетическая энергия (эВ);
-
кинетическая температура (К);
-
coul1 и coul2 – действительная и мнимая часть Эвальдовой суммы (выводится только при использовании ключевого слова pme в электростатике);
-
полная кулоновская энергия (эВ) – если используется электростатика. В случае Эвальда – это сумма coul1, coul2 и «постоянного» вклада;
-
P+K (эВ) – сумма потенциальной и кинетической энергии (выводится только для излучательного термостата);
-
термальная энергия (эВ) – только для излучательного термостата, сумма H по всем атомам;
-
полная энергия (эВ) – сумма всех энергий;
-
энергия связей (эВ) – только если заданы валентные связи;
-
энергия углов (эВ) – только если заданы валентные углы;
-
энергия во внешнем поле (эВ) – только если задано внешнее электрическое поле (директива elecField);
-
далее 6 колонок – импульс, переносимый через каждую из шести стенок бокса накопительным итогом, в эВ * пс / Å;
-
давление (атм). Давление рассчитывается как производная импульса (см. выше) по времени приведенная к площади стенки бокса, усредненная по всем 6 стенкам бокса;
-
далее идут колонки с количеством атомов каждого типа, если их число изменяется.
5.3 Файл rdf.dat – содержит радиальные функции распределения для всех пар типов атомов, первая колонка – расстояние в Å.
5.4 Файл rdf_n.dat – аналог файла rdf.dat, но не по типам атомов, а по их остовам. Например, если у вас несколько подтипов атомов кислорода, но для вывода радиальных функций они должны считаться неразличимыми, можете задать им один остов (раздел spec, файла field). Выводится, если в конце директивы rdf указано слово nucl.
5.5 Файл msd.dat – первые две колонки время (пс) и номер шага. Затем по 6 колонок для каждого типа – количество атомов, прошедших через одну из 6 стенок бокса накопительным итогом.
5.6 Файл velocities.dat – для каждого типа частиц перечень скоростей и их проекций на оси Ox, Oy и Oz. Скорости даны в Å/пс.
5.7 Файл traj.dat – «траектории движения». Первые две колонки – время (пс) и номер шага. Затем координаты x и y для каждого из выводимых атомов. В шапке координаты x указан также тип атома. Обратите внимание, что непостоянное поле сил позволяет менять тип частиц, в шапке будет указан тип на начальный момент времени.
5.8 Файл length.dat – перечень длин связей каждого типа. Только при использовании связей.
5.9 Файл stat_bnd.dat – Статистика по связям. Только при использовании связей. Первые две колонки – время (пс) и номер шага, затем общее число связей, затем для каждой связи – количество, средняя длина (Å) и среднее время жизни (пс).
5.10 Файл tchar.dat – только для излучательного термостата. Тепловая энергия и «радиус» для каждого типа атома (см. подробнее «Излучательный термостат»).
5.11 Файл nCN.dat – распределение по координационным числам. Вывод активируется разделом ncn в файле control.txt. Первая колонка – координационное число, затем в количество пар частиц с указанным координационным числом. Результат вычисляется для финальной конфигурации системы, не усредняется по шагам.
6 Особенности
Теперь подробнее об оригинальных фичах программы, которые выходят за пределы классической молекулярной динамики.
6.1 Связывание
Можно задать динамическое образование связей/углов прямо в ходе моделирования. Либо использовать эту особенность, чтобы задать все связи/углы в системе, а затем уже запустить классическую молекулярную динамику. Для того, чтобы указать, что частицы A и B оказавшись на расстоянии до N ангстрем образовали связь нужно сделать соответствующие объявления в файле field.txt. Чтобы избежать повторного связывания одной и той же пары атомов, введен массив parents, где хранятся индексы атомов, связанных с данным валентной связью (или -1, если у атома нет связей). Проверка выглядит следующим образом:
if (parents[i] != j && parents[j] != i)
где i и j – индексы атомов. Элемент массива parents[i] перезаписывается каждый раз, когда атом с индексом i образует связь, поэтому может получится, что проверка не спасёт от повторного связывания. Однако, можно показать, что для систем, где атомы с произвольным числом связей связаны друг с другом через атомы, имеющие две связи, этой проверки достаточно. К таким системам относятся, например, все оксиды и сульфиды. Действительно, введем следующие типы атомов: Of, O1, O2 – свободный кислород, 1- и 2-связанный. Зададим, что Of образует связь с превращением в O1, а O1 после связывания становится O2. O2 уже не образует связей, а у Of их нет – т.е. для них проверки не актуальны. Частица O1 имеет только одну связь и значение parents для неё определено однозначно.
Рассмотрим пример. Пускай хотим смоделировать раствор слабой кислоты (скажем плавиковой) в воде. Здесь и далее использованы произвольные потенциалы, плотность и т.д. просто для демонстрации возможностей программы. Воспользуемся генератором:
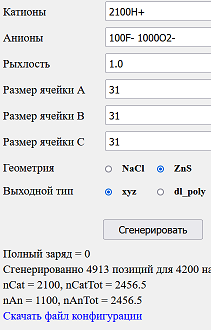
Возьмём 100 молекул плавиковки и 1000 воды: итого 2100 протонов, 100 ионов фтора и 1000 ионов кислорода. Вспомнив, что 6*1023 молекул воды при стандартных условиях занимают 18 см3 (18*1024 Å3), можно приблизительно оценить, что наши 1000 молекул будут занимать 1000/(6 * 1023) * 18*1024 = 30000 Å3, это куб со стороной около 31 Å. В результате работы генератора получим периодическую (кристаллическую) систему и возьмём её в качестве отправной точки.

Кроме этого файла с координатами атомов потребуются ещё 3 (cuda.txt, control.txt, field.txt). Файл cuda.txt, можете просто скопировать содержимое, поправив значение singproc под ваше количество ядер в мультипроцессоре:
cuda.txt
multproc 0
singproc 128
nthread a 16
nthread b 32
bindtraj threads 1 32
nstep stat 50
nstep msdstat 50
nstep bondstat 50
nstep traj 10
nstep bindtraj 20Файл control.txt:
timestep 0.001
nstep 10000
nequil 5000
eqfreq 200
temperature 1200.0 nose 0.2
init_vel gaus
permittivity 1.0
elec fenn 8.0 0.4
cell_list 2.7
max_neigh 185
rdf 8.0 0.02 10 200000 nucl
stat 200Традиционно используется величина шага 0.001 пс. Число шагов (nstep) и число уравновешивающих шагов (nequil) задайте произвольно. Температура здесь 1200, чтобы побыстрее перемешать атомы и скорее все кислороды наткнулись на протоны с образованием связей. Электростатику приказали считать по Феннелю, с радиусом обрезания 8 ангстрем и константой α = 0.4. Это раза в два быстрее, чем методом Эвальда и не надо думать, сколько k-векторов выбрать. Попробуйте поиграть с параметром cell_list (размер субячейки бокса, в ангстремах), чтобы получить максимальную скорость.
Самый интересный файл — файл field.txt:
field.txt
spec 7
O2- O 16.0 -2.0 0.0
Ot O 16.0 -1.6 0.0
Oc O 16.0 -1.2 0.0
H H 1.0 1.0 0.0
Hb H 1.0 0.6 0.0
F- F 19.0 -1.0 0.0
Fb F 19.0 -0.6 0.0
vdw 28
O2- H buck 6.0 150.0 0.3 0.0
Ot H buck 6.0 150.0 0.3 0.0
Oc H buck 6.0 150.0 0.3 0.0
O2- Hb buck 6.0 150.0 0.3 0.0
Ot Hb buck 6.0 150.0 0.3 0.0
Oc Hb buck 6.0 150.0 0.3 0.0
O2- F- buck 6.0 1000.0 0.3 0.0
Ot F- buck 6.0 1000.0 0.3 0.0
Oc F- buck 6.0 1000.0 0.3 0.0
O2- Fb buck 6.0 1000.0 0.3 0.0
Ot Fb buck 6.0 1000.0 0.3 0.0
Oc Fb buck 6.0 1000.0 0.3 0.0
O2- O2- buck 6.0 1000.0 0.3 0.0
O2- Ot buck 6.0 1000.0 0.3 0.0
O2- Oc buck 6.0 1000.0 0.3 0.0
Ot Ot buck 6.0 1000.0 0.3 0.0
Ot Oc buck 6.0 1000.0 0.3 0.0
Oc Oc buck 6.0 1000.0 0.3 0.0
F- H buck 6.0 100.0 0.3 0.0
F- Hb buck 6.0 100.0 0.3 0.0
Fb H buck 6.0 100.0 0.3 0.0
Fb Hb buck 6.0 100.0 0.3 0.0
F- F- buck 6.0 1000.0 0.3 0.0
F- Fb buck 6.0 1000.0 0.3 0.0
Fb Fb buck 6.0 1000.0 0.3 0.0
H H buck 6.0 1000.0 0.3 0.0
H Hb buck 6.0 1000.0 0.3 0.0
Hb Hb buck 6.0 1000.0 0.3 0.0
bonds 3
1 Ot Hb harm 20.0 0.9 con con
2 Oc Hb harm 20.0 0.9 con con
3 Fb Hb mors 7.0 1.5 1.0 7.0 con br 1.2 F- H
angles 1
1 Oc hcos 10.0 -0.829
linkage 3
O2- H 2.0 1
Ot H 2.0 2
F- H 2.0 3
angle_forming 1
Oc 1Здесь заданы 7 типов атомов (пока у нас присутствуют 3: O2-, H и F), далее увидим откуда берутся остальные. Обозначим кислород с одной связью как Ot (terminal) и с двумя – как Oc (chain). Также введем обозначения для связанных фтора и водорода как Hb и Fb (bonded). Массы укажем табличные, а заряды возьмём так, чтобы связанные аналоги частиц имели меньший заряд и соблюдалась электронейтральность.
Далее идёт перечисление парных потенциалов для всех возможных пар частиц. Потенциалы взяты произвольно в форме потенциала Букингема с радиусом обрезания 6 Å, для пар частиц противоположного заряда отталкивание задано послабее.
Раздел bonds описывает 3 типа связи. Первые два между разными формами кислорода и водородом, в форме гармонического потенциала и не изменяющиеся при сжатии/растяжении (директивы con). Последний тип связи – между фтором и водородом в форме потенциала Морзе и рвётся по достижении длины 1.2 Å с преобразованием частиц (Fb → F-, Hb → H), что эмулирует диссоциацию молекулы кислоты. Изначально ни одной из этих связей в системе не присутствует, для их образования служит раздел linkage.
В разделе linkage сказано, что частицы O2- и H оказавшись на расстоянии до 2х Å образуют связь № 1 из раздела bonds, при этом O2- → Ot, H → Hb (программа ставит в соответствие первый тип атома в разделе bonds с первым типом атома в разделе linkage, поэтому следите, чтобы кислород превращался в кислород и т.д.!) Аналогичным образом связываются ещё 2 пары частиц. Таким образом, в системе будут появляться частицы Ot, Oc, Hb и Fb, отсутствующие изначально. А раздел angle_forming говорит о том, что как только появится частица Oc будет создан валентный угол с вершиной в нём (боковые атомы определяются на основе имеющихся связей) и потенциалом валентного угла из раздела angles.
Прогоним систему разок и бОльшая часть атомов свяжется. Повторим процедуру для связывания остальных атомов: среди выходных файлов есть revcon.xyz, revbonds.txt и revangles.txt. Переименуем их в atoms.xyz, bonds.txt и angles.txt и не забудем добавить в файл field.txt директивы:
bond_list 1
angle_list 1которые означают, что нужно считывать списки связей и углов. Повторив процедуру несколько раз, мы в конце концов свяжем весь кислород и получим картинку с характерными уголковыми молекулами воды:

Теперь можно упростить файл field.txt. Связи O-H все образованы и диссоциацией воды мы пренебрегаем, так что часть директив можно удалить. Более того, у нас не осталось частиц O2-, так что можно убрать их из раздела spec и удалить соответствующие парные потенциалы. Можно было бы из частиц убрать и тип Ot, но он, к сожалению, фигурирует в связях, причем в первой и, если её убрать нарушится индексация в файле bonds.txt. На будущее – старайтесь ненужные связи ставить последними.
Новый файл field.txt
spec 6
Ot O 16.0 -1.6 0.0
Oc O 16.0 -1.2 0.0
H H 1.0 1.0 0.0
Hb H 1.0 0.6 0.0
F- F 19.0 -1.0 0.0
Fb F 19.0 -0.6 0.0
vdw 15
Oc H buck 6.0 150.0 0.3 0.0
Oc Hb buck 6.0 150.0 0.3 0.0
Oc F- buck 6.0 1000.0 0.3 0.0
Oc Fb buck 6.0 1000.0 0.3 0.0
Oc Oc buck 6.0 1000.0 0.3 0.0
F- H buck 6.0 100.0 0.3 0.0
F- Hb buck 6.0 100.0 0.3 0.0
Fb H buck 6.0 100.0 0.3 0.0
Fb Hb buck 6.0 100.0 0.3 0.0
F- F- buck 6.0 1000.0 0.3 0.0
F- Fb buck 6.0 1000.0 0.3 0.0
Fb Fb buck 6.0 1000.0 0.3 0.0
H H buck 6.0 1000.0 0.3 0.0
H Hb buck 6.0 1000.0 0.3 0.0
Hb Hb buck 6.0 1000.0 0.3 0.0
bonds 3
1 Ot Hb harm 20.0 0.9 con con
2 Oc Hb harm 20.0 0.9 con con
3 Fb Hb mors 5.0 1.5 1.0 5.0 con br 1.05 F- H
angles 1
1 Oc hcos 10.0 -0.829
linkage 1
F- H 2.0 3
bond_list 1
angle_list 1Поправим температуру на комнатную в файле control.txt и запускаем. Затем можно посмотреть, как осциллирует количество связей HF со временем (файл bnd_stat.dat) и оценить константу диссоциации. А можно наоборот: корректировать потенциалы так, чтобы воспроизвести экспериментальные данные по диссоциации.
Факультативная задача. Водородные связи.
Без рассмотрения водородных связей предыдущая задача выглядит неполной. Программа aztotmd сконструирована, в том числе, для учета водородных связей. Алгоритмические отличия: при образовании водородной связи не перезаписывается массив parents, что позволяет обойти ограничение в две связи на кислород (см. начало раздела). Кроме того, при образовании водородных связей не образуются валентные углы. У атома водорода в сумме может быть две связи включая водородные и обычные. Таким образом, у нас появляются ещё 3 частицы, назовём их Hbh, Hh и Hhh: протон с одной обычной связью и одной водородной, водород с одной водородной связью и водород с двумя водородными связями. Каждый из этих 3х типов водорода может быть связан водородной связью с тремя типами самых электроотрицательных элементов: Oc, F- и Fb, итого 9 новых типов связей, возьмём их в форме потенциала Морзе. Зададим, что все эти связи распадаются при достижении длины 2 Å. Кроме того, зададим, что водородная связь Hh…F- превратится в обычную ковалентную Hb-Fb при сокращении до 1.05 Å. Кроме того, поскольку уже связанные водороды (Hb) могут превратиться в Hbh нужно ввести обычные связи с водородно-связанными атомами: Fb-Hbh и Oc-Hbh. В разделе h-bonds указываем какие связи считать водородными и какой атом с связи считать атомом водорода.
Теперь заметим, что у нас есть два типа связи между одними и теми же парами частиц,
одна из них водородная, а другая – обычная (связи 4 и 12 между Oc и Hbh и связи 5 и 14 между Fb и Hbh). Когда Hb превращается в Hbh связь 2 должна превращаться в 4, а 3 – в 5. Фтор может менять своё состояние с F- на Fb, при этом если он был связан водородной связью (№ 13), то та должна изменится на 14, а не на 5. Задать это поведение можно используя раздел evol_bonds.
Файл field.txt:
spec 9
Ot O 16.0 -1.6 0.0
Oc O 16.0 -1.2 0.0
H H 1.0 1.0 0.0
Hb H 1.0 0.6 0.0
F- F 19.0 -1.0 0.0
Fb F 19.0 -0.6 0.0
Hbh H 1.0 0.6 0.0
Hh H 1.0 1.0 0.0
Hhh H 1.0 1.0 0.0
vdw 36
Oc H buck 6.0 150.0 0.3 0.0
Oc Hb buck 6.0 150.0 0.3 0.0
Oc F- buck 6.0 1000.0 0.3 0.0
Oc Fb buck 6.0 1000.0 0.3 0.0
Oc Oc buck 6.0 1000.0 0.3 0.0
F- H buck 6.0 100.0 0.3 0.0
F- Hb buck 6.0 100.0 0.3 0.0
Fb H buck 6.0 100.0 0.3 0.0
Fb Hb buck 6.0 100.0 0.3 0.0
F- F- buck 6.0 1000.0 0.3 0.0
F- Fb buck 6.0 1000.0 0.3 0.0
Fb Fb buck 6.0 1000.0 0.3 0.0
H H buck 6.0 1000.0 0.3 0.0
H Hb buck 6.0 1000.0 0.3 0.0
Hb Hb buck 6.0 1000.0 0.3 0.0
Hbh Oc buck 6.0 150.0 0.3 0.0
Hbh F- buck 6.0 1000.0 0.3 0.0
Hbh Fb buck 6.0 1000.0 0.3 0.0
Hbh H buck 6.0 1000.0 0.3 0.0
Hbh Hb buck 6.0 1000.0 0.3 0.0
Hh Oc buck 6.0 150.0 0.3 0.0
Hh F- buck 6.0 1000.0 0.3 0.0
Hh Fb buck 6.0 1000.0 0.3 0.0
Hh H buck 6.0 1000.0 0.3 0.0
Hh Hb buck 6.0 1000.0 0.3 0.0
Hhh Oc buck 6.0 150.0 0.3 0.0
Hhh F- buck 6.0 1000.0 0.3 0.0
Hhh Fb buck 6.0 1000.0 0.3 0.0
Hhh H buck 6.0 1000.0 0.3 0.0
Hhh Hb buck 6.0 1000.0 0.3 0.0
Hbh Hbh buck 6.0 1000.0 0.3 0.0
Hbh Hh buck 6.0 1000.0 0.3 0.0
Hbh Hhh buck 6.0 1000.0 0.3 0.0
Hh Hh buck 6.0 1000.0 0.3 0.0
Hh Hhh buck 6.0 1000.0 0.3 0.0
Hhh Hhh buck 6.0 1000.0 0.3 0.0
bonds 14
1 Ot Hb harm 20.0 0.9 con con
2 Oc Hb harm 20.0 0.9 con con
3 Fb Hb mors 7.0 1.5 1.0 7.0 con br 1.05 F- H
4 Oc Hbh harm 20.0 0.9 con con
5 Fb Hbh mors 7.0 1.5 1.0 7.0 con br 1.05 F- Hh
6 Oc Hh mors 0.022 1.0 1.7 0.022 con br 2.0 Oc H
7 F- Hh mors 0.05 1.0 1.7 0.05 mut 1.05 3 br 2.0 F- H
8 Fb Hh mors 0.05 1.0 1.7 0.05 con br 2.0 Fb H
9 Oc Hhh mors 0.022 1.0 1.7 0.022 con br 2.0 Oc Hh
10 F- Hhh mors 0.05 1.0 1.7 0.05 con br 2.0 F- Hh
11 Fb Hhh mors 0.05 1.0 1.7 0.05 con br 2.0 Fb Hh
12 Oc Hbh mors 0.022 1.0 1.7 0.022 con br 2.0 Oc Hb
13 F- Hbh mors 0.05 1.0 1.7 0.05 con br 2.0 F- Hb
14 Fb Hbh mors 0.05 1.0 1.7 0.05 con br 2.0 Fb Hb
angles 1
1 Oc hcos 10.0 -0.829
linkage 10
F- H 1.1 3
Oc H 2.0 6
F- H 2.0 7
Fb H 2.0 8
Oc Hh 2.0 9
F- Hh 2.0 10
Fb Hh 2.0 11
Oc Hb 2.0 12
F- Hb 2.0 13
Fb Hb 2.0 14
h-bonds 9
6 Hh 7 Hh 8 Hh 9 Hhh 10 Hhh 11 Hhh 12 Hbh 13 Hbh 14 Hbh
evol_bonds 3
2-4 3-5 13-14
bond_list 1
angle_list 16.2 Излучательный термостат и температуро-зависимое поле сил
Излучательный термостат возник из сомнений в том, что температура – это величина пропорциональная среднекинетической энергии частиц (кинетическая температура). Излучательный термостат построен на законе излучения абсолютно-черного тела (которое зависит только от температуры). Термостат основан на следующих предположениях:
-
излучают атомы;
-
атомы находятся в состоянии теплового возбуждения;
-
величина теплового возбуждения различна для разных атомов (поскольку тепловое излучение не монохромное);
-
энергия теплового возбуждения запасается в виде увеличения расстояния от ядра до максимума электронной плотности (Боровский радиус).
Подробнее реализация излучательного термостата описана описана здесь. Используйте ключевое слово radi для активации излучательного термостата. Приятным бонусом к излучательному термостату идёт возможность использования температуро-зависимого поля сил, как было обещано в ответ на этот комментарий. Температуро-зависимый потенциал зависит от температуры неявно, через Боровские радиусы, и представлен в форме:

где С1, С2, a и b – константы, ri, rj – Боровские радиусы i-ой и j-ой частиц. Для использования этого потенциала необходимо задать секцию radii в файле field.txt. Сам потенциал вызывается ключевым словом surk, параметры перечисляются в следующем порядке: С1, С2, a, b. Пример файла field.txt при использовании температуро-зависимого потенциала.
spec 2
Na Na 23.0 0.3 0.0
Cl Cl 35.4 -0.3 0.0
vdw 4
Na Cl surk 6.0 75.0 8.0 1.0 1.0
Cl Na surk 6.0 75.0 8.0 1.0 1.0
Na Na surk 6.0 4000.0 260.0 1.0 1.0
Cl Cl surk 6.0 1500.0 450.0 1.0 1.0
radii 2
Na 27.3 47.31 0.2
Cl 37.8 47.31 0.2Поскольку указанный потенциал становится несимметричным, относительно перестановки частиц i и j, то для каждой пары частиц разного типа необходимо указывать два потенциала: A…B и B…A.
Предложенная схема позволяет объяснить ряд явлений:
-
термическое расширение (растет Боровский радиус);
-
увеличение сжимаемости с температурой (размер электронных оболочек увеличивается => электронная плотность уменьшается, следовательно уменьшается отталкивание при одной и той же степени перекрывания оболочек);
-
образование дефектов (часть атомов обладает достаточным термическим возбуждением);
-
плавление (из-за разных радиусов набирается достаточно ассиметрии, чтобы разрушить кристалл).
Излучательный термостат справляется и с обратной задачей: кристаллизацией разупорядоченной фазы при снижении температуры. Численные эксперименты показали, что система из одинаковых атомов при этом стремится кристаллизоваться в ГЦК-решётку, а из двух противоположно-заряженных в Fm3m, в которой кристаллизуются большинство галогенидов щелочных металлов и многие другие бинарные соединения (LiF, LiCl, LiBr, LiI, NaF, NaCl, NaBr, NaI, KF, KCl, KBr, KI, CsF, MgO, CaO, FeO, β-NiO, CdO, MgS, CaS и т.д.). На рисунке ниже представлен результат охлаждения бинарного расплава, видно образование упорядоченных областей, повернутых разными плоскостями, т.е. излучательный термостат также воспроизводит зернообразование.

7 Модификация программы
Этот раздел для тех, кто хочет модифицировать программу.
7.1 Добавление парных потенциалов
Для чтения парного потенциала необходимо поправить файлы vdw.h и vdw.cpp. Функция read_vdw считывает парный потенциал из файла field.txt. Константы:
-
nVdWType– количество парных потенциалов; -
массив
vdw_abbr– ключевые слова, обозначающие парный потенциал; -
массив
vdw_param– количество параметров парных потенциалов; -
массивы
vdw_scale0..4– масштабирующие коэффициенты для параметров парных потенциалов (единицы длины следует масштабировать коэффициентом r_scale, энергии – E_scale).
Далее в модуле cuVdW.cu следует описать функции, вычисляющие значение парного потенциала. Обычно описываются 4 функции на один потенциал, вычисляющие: только энергию, только энергию по известному расстоянию, силу и энергию, силу и энергию по известному расстоянию. Для температуро-зависимых потенциалов задается одна функция, вычисляющая энергию и силу. В функции define_vdw_func следует дописать присваивание указателей на написанные функции.
7.2 Добавление новых вычисляемых параметров
Добавьте нужную переменную в структуру cudaMD (cuStruct.h). Соответствующие расчеты добавьте в функцию calc_quantities (main.cu), обращаясь к переменным через md->.
7.3 Добавление параметров в выводимую статистику
Чтобы добавить параметр в выводимую статистику нужно поправить до 5 функций: init_cuda_stat, prepare_stat_addr, start_stat, end_stat и read_cuda и, возможно, структуру hostManagMD (cuStruct.h):
-
init_cuda_stat(cuStat.cu) определяет размер буфера статистики, типы данных хранимых в каждом элементе буфера (0 – int, 1 – float), сдвиги статистик разного типа относительно адреса размещения буфера; -
prepare_stat_addr(cuStat.cu) задает адреса переменных, которые копируются в буфер статистики; -
start_stat(cuStat.cu) открывает файлы для записи статистики, записывает заголовки; -
end_stat(cuStat.cu) копирует остатки информации из буфера на хост, закрывает файлы; -
read_cuda(cuInit.cu) – поправьте эту функцию, если вводите новый тип статистики и нужно считать размер буфера для этой статистики из файла cuda.txt.
8 Предупреждения / сообщения об ошибках
Программа выдает 3 типа сообщений, если что-то пошло не так:
-
WARNING[NNN] – возможно неточность во входных данных: переопределили парный потенциал, забыли указать потенциалы, забыли задать заряды частиц и т.д. Не мешает работе программы, внимательно прочитайте и убедитесь, что эффект соответствует вашим ожиданиям.
-
ERROR[NNN] – ошибка. Дальнейший расчет невозможен или приведёт к некорректному результату. Поправьте и перезапустите.
-
UBEH[NNN] – неожиданное поведение. Скорее всего что-то неправильно задано, перепроверьте все введенные данные и перезапустите программу.
PS
Приветствуются различные комментарии и уточнения. Также принимаю заявки на добавление функционала.


