Введение
«Предрассветная дымка нехотя отступала по оврагам, проступали стебли ржи, переливающиеся под взмахами ветра. Птицы уже успели обрадоваться утру и ненавязчиво щебетали над ухом. Последние капли сна упали в чашку ароматного кофе. Приятно встречать диск солнца, растягивая застывшие суставы и вглядываясь в даль. Кто это? Застыла мысль, когда взгляд скользнул на тропинку, бегущую из леса. Широкая улыбка озарила лицо. С первых движений он узнал ее. Только она могла двигаться с такой грацией и изяществом лани. Рука замерла на полпути к столу. Продолжая улыбаться, он вдруг резко повернулся и зашагал на кухню. Появились на столе еще одна чашка и поднос с ягодами. Аромат лавандового сиропа заполонил веранду. Хороший будет день, подумалось ему, приятный завтрак – уж точно.
Горсть малины быстро исчезала с подноса ягода за ягодой. Нежный женский голос рассказывал последние вести. За прошедшую неделю не поставили ни одного креста на городском кладбище. Мы дожили! — Выдох радости вырвался из уже скукоженных от возраста легких. Да!- ответила она ему. Эти чертовы четверть века. Четверть века, которые не оставили на твоем лице ни одного гладкого участка.
Его уже глубоко близорукие глаза смотрели на нее и в них она видела бесконечные линии графиков масс-спектрометра, видела ту бездонную усталость, которая не отступала в попытках уложить его в ящик. Он справился, выдержал. У людей больше не было ужасного животного страха сгинуть, наверное, самой ужасной смертью, что можно было представить…»
Вот такое начало фантастического рассказа прочитал я на досуге. В нем описывается новый вид биологического оружия. Ужасающий в своей разрушительной мощи. Людей охватывало оцепенение, когда они узнавали свою участь. Страх перед этой невидимой и неотвратимой напастью был хуже самой смерти.
«Почесуха»
Великобритания, первая половина 18 века. Туман рассеивается над зелеными сочными полями. Большое стадо овец не спеша продвигается в сторону реки. Вдруг мы замечаем что-то необычное. Как минимум пятая часть овец неистово катается по траве и трется шкурой о камни, выступающие над поверхностью земли, оставляя за собой беспорядочные клочья. Расчесанные, потерявшие шерсть бока покрыты страшными язвинами и эррозиями. Часть овец уже не способна чесаться, они просто медленно, трясущейся походкой, скрежеща зубами идут по полю, к месту последнего успокоения. Что же это за напасть такая, думали скотоводы, называя болезнь по ее главному проявлению – СКРЕПИ («почесуха»). Эта зараза не отступала столетиями, то и дело появляясь то там, то здесь, оставляя после себя разоренные семьи.
«Мозгоедки»
Настоящие ученые – не совсем обычные люди, знакомясь с их биографиями, часто поражаешься, каким же сумасшедшим вихрем крутилась шарманка судьбы.
Одна из ниточек нашего повествования начнется с жизнеописания Даниэля Карлтона Гайдусека (1923-2008). Представим себе молодого человека, ему 23 года, он только что получил степень магистра в Гарварде, с огромным воодушевлением едет работать в Калифорнийский технологический, да не, а бы с кем, а с самим Лайнусом Полингом (дважды Нобелевский лауреат). Спустя три года он принимает приглашение и занимает должность научного сотрудника факультета педиатрии и инфекционных болезней уже в своей альма-матер. Несмотря на столь успешную карьеру, что-то не ладится и не дает ему покоя. Не проработав и 3-х лет бросает все и уезжает сначала в Тегеран в институт Пастера, а через три года странным зигзагом через Гиндукуш, оказывается в медицинском институте Уолтера и Элизы Холл в Мельбурне. Дауншифтинг, не иначе.
Именно в Асвтралии, состоялось судьбоносное знакомство Даниэля Гайдусека с медицинским работником Винсентом Зигасом (1920-1983), который тесно общался с племенами Папуа Новой Гвинеи, оказывая им медицинскую помощь. Зигас рассказывает Даниэлю о неизвестной болезни, странные симптомы которой проявляются у единственного народа – Форе. Гайдусек в нетерпении бросается изучать язык аборигенов и спустя несколько месяцев Зигас привозит и представляет Гайдусека народности Форе. Почти год они живут среди дикого племени, отслеживая все привычки, обычаи. Наблюдают больных и проводят вскрытия погибших. [1]
Вот как они описывают последовательность развития столь заинтересовавшей их болезни в своей статье:
«… Человека настигает апатия и непреодолимая усталость. Спустя месяц или чуть больше начинаются характерные подергивания и подрагивания. Все более отчетливым и постоянным становится тремор конечностей, туловища и головы. Человек теряет способность передвигаться. В срок от года до двух лет наступает смерть. Члены племени Форе называют эту болезнь «Куру», что означает дрожь, порча. И считают, что причина кроется в сглазе шамана».
После проведения вскрытия, у многих погибших от болезни Зигас и Гайдусек обнаруживали превращения мозга в губчатую субстанцию. [2]
Длительное проживание внутри племени позволило Гайдусеку и Зигасу обнаружить причину развития болезни. Оказалось, что племя Форе практиковало каннибализм.
После смерти одного из старших членов рода его тело разделывали, вскрывали черепную коробку и съедали мозг, так как считалось, что поедание головного мозга представляет собой ритуал последних почестей умершему, а тот, кто съест мозг, приобретёт его мудрость, смелость и остальные благородные качества, которыми он владел. Обычно большую часть мозга съедали женщины и потому среди них число заболевших было выше.[3] С искоренением столь пагубного обычая практически полностью была побеждена и болезнь «куру».
За описание болезни «куру» в 1976 году Гайдусек получил Нобелевскую премию. И тут не ясны мотивы Нобелевского комитета, который обошел вниманием Винсента Зигаса. В своей Нобелевской лекции Гайдусек рассказывал про вирусную природу болезни «куру». Узнаем прав он был или нет чуть позже.

Больной, пораженный «куру».
«Успел»
Пока же перенесемся в Германию. Начало 20 века, психиатрическая клиника в Бреслау, кафедра под началом Алоиза Альцгеймера. На работу приходит молодой судовой врач, который решил стать неврологом. Пока он упорно постигает азы профессии ему удается обнаружить пациентов с никому дотоле неизвестным заболеванием. Исследования прерывает Первая мировая война, которая вернула доктора Ганса-Герхарда Крейтцфельда в состав военно – морского флота. Только в 1920 году, спустя 6 лет, он публикует описание болезни.
В описании обнаруживаем, что пациенты с высокой скоростью теряли память, переставали осознавать себя и через 8-12 месяцев после первых проявлений клинической картины умирали. В препаратах мозга, полученных от таких пациентов, были обнаружены характерные «губчатые структуры».
Стоит сказать, что ему несказанно повезло, опоздай он еще на полгода, и ветер времени развеял бы его имя в веках, так как спустя несколько месяцев выходит работа Альфонса Якоба с описанием той же самой болезни, которая обрела имя своих открывателей – болезнь Крейтцфельда – Якоба(БКЯ).
Что может быть общего между скрепи, куру и БКЯ? Именно такой вопрос начали задавать ученые к 50-м годам 20 века, ведь эти заболевания были так похожи длинным инкубационным периодом в 5 – 10 лет и неизменной печальной судьбой пораженного, будь то животное или человек. Причем повреждения прежде всего настигали мозг. Так и назвали эту группу заболеваний нейродегенерации с длительным инкубационным периодом.
Основная часть
Эксперименты
С развитием экспериментальных методов биохимии стало возможным наконец подступиться к этим патологиям. Неимоверно сложно было искать источник заражения при условии, что проявления болезни можно обнаружить только спустя годы, несмотря на трудности, попытки выяснить причины болезни не прекращались. В лабораториях искали способы упростить экспериментальную работу, сократив срок инкубационного периода до приемлемого.
Так Пэтисон и Гуили смогли передать болезнь от овечки к овечке с помощью бесклеточных фильтратов. Для начала лабораторных экспериментов оставался один шаг – передать болезнь от овцы к лабораторному животному. И его делает Чандлер в 1960 году, о чем пишет небольшую, но очень известную статью в 1961 году[4]. Ему удалось заразить лабораторную мышь с помощью вещества из мозга больного животного. Причем в последних исследованиях проявление заболевания пришлось ждать всего 7 месяцев. Стало удобно исследовать болезнь в лабораторных условиях.
Активизировались поиски инфицирующего агента. Установить его долгое время не удавалось. Сначала искали неизвестный вирус, похожий на герпес или энцефалит, но ничего не находили. Всех исследователей удивляло, что способность к заражению у этой субстанции, выделенной из мозга больных животных, сохранялась и после сильного длительного нагрева, и после обработки ацетиэтилениминам. Были поставлены эксперименты, в которых фильтрат подвергли обработке жестким УФ и ионизирующим излучением. Несмотря на это, фильтрат сохранил способность к заражению.[5] Стали закрадываться подозрения, что вирусы в этом случае не причем, ведь нуклеиновые кислоты (непременная составляющая любого вируса) при таком воздействии попросту разрушаются.
Гриффит в небольшой заметке на полторы страницы текста в 1967 году озвучивает еретическую мысль – инфекционный агент не содержит нуклеиновых кислот.[6] Это белок, который способен к самовоспроизводству в клетке. Именно с этой заметки началась новая эра.
Инфекционный белок
Эксперименты по исследованию скрепи все также оставались сложными и длительными. Только спустя 15 лет, Стенли Прузинер в калифорнийском университете Сан-Франциско выделил и описал агент, способный в чистом виде вызывать развитие болезни скрепи. Выяснилось, что это удивительное вещество устойчиво к нагреванию, сохраняет инфекционность после обработки различными повреждающими агентами, такими как: протеиназа К, мочевина, гуанидинхлорид, детергентыи, SDS и нуклеазы — ферменты повреждающие ДНК, но было также обнаружено, что данный инфекционный агент чувствителен к ионизирующему излучению в присутствии кислорода, что характерно для гидрофобных белков имеющих большое сродство к липидам. [8]
Прузинер придумал название для агента, вызывающего скрепи – «ПРИОН» (prion –proteinacious infectious particle). Прионный белок (Prione Protein PrP) был выделен чуть позже. Методы секвенирования в то время уж были развиты достаточно хорошо и быстро позволили установить первичную последовательность PrP. Все начали искать источник PrP. Статья в Nature от 1985 года, ознаменовавшая собой окончание поисков, поставила многих исследователей в тупик: матричная РНК (молекула – шаблон по которой потом синтезируются белки) необходимая для синтеза PrP обнаружилась в здоровом мозге. [7]
Это означало только одно — белок, ответственный за развитие болезни всегда присутствует в головном мозге, не зависимо от развития болезни. Позже выяснили, что ген, кодирующий PrP есть у всех млекопитающих, а также у птиц и рыб.
Расположение отдельных участков 20-й хромосомы человека с отметкой места нахождения гена, кодирующего PrPС.
Структура белка
Что же это за удивительный белок? Функция его и спустя 37 лет с момента обнаружения не выяснена (тут стоит сказать спасибо западной модели грантово-статейной науки). Известно, что этот белок связан с клеточной мембраной. И возможно отвечает за межклеточные взаимодействия в мозге.
Чтобы понять, как же обычный белок становится заразным необходимо обратиться к структуре белков. Первичная структура белка – это последовательность аминокислотных остатков. Эта последовательность и у нормального PrPC и у инфекционной формы PrPSc одинаковая.
Отличия удалось обнаружить на уровне вторичной и третичной пространственной структуры. У PrPC вторичная структура представлена 42% α-спиралей и 3% β-структур, а в тоже время PrPSc содержит 30% α-спиралей и 43% β-структур. Этот факт позволил предположить, что патологическая форма белка образуется при неправильном сворачивании аминокислотной последовательности в β-складчатые слои.
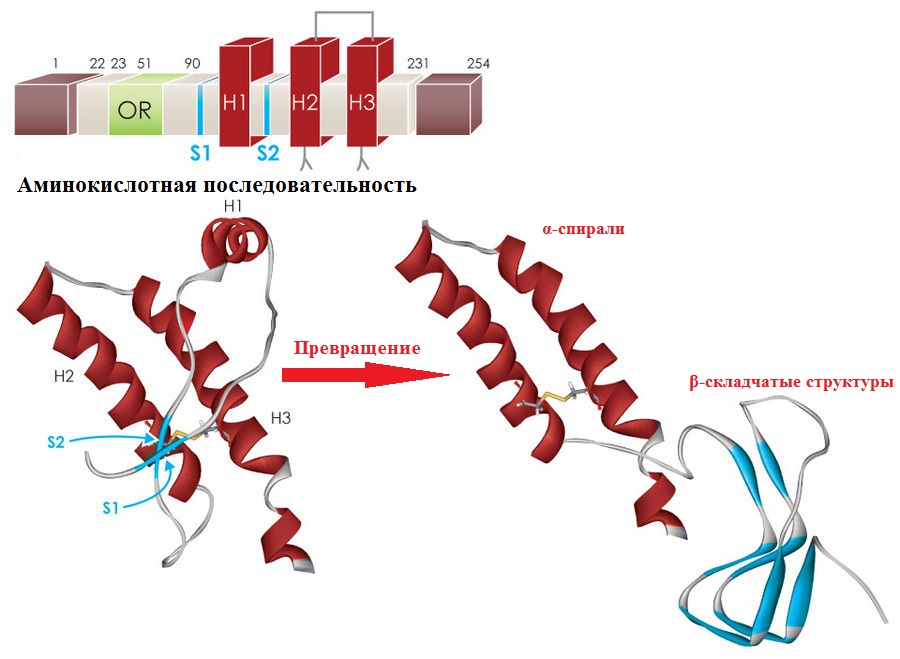
На изображении вверху аминокислотная последовательность PrP, с выделенными участками различных белковых структур H1,H2, Р3 – α спирали. Внизу показано превращение спиралей в β-складчатые слои. Изображение: By Olivia May, Ph.D
Прионная гипотеза
На основе накопленных данных в 1991 году Прузинер формирует «Прионную гипотезу», в которой постулирует следующее:
• инфекционным агентом является белок PrPSc,
• инфекционный агент PrPSc может реплицировать себя в отсутствие нуклеиновой кислоты,
• превращение белка из нормальной формы (PrPC) в инфекционную (PrPSc) происходит путем конформационного перехода,
• конформационный переход PrPC в PrPSc может происходить спонтанно, приводя к спорадическим формам прионных болезней. Он может быть вызван поступлением в организм патологической формы PrPSc извне (приобретенные формы прионных заболеваний).
• переход может произойти из-за мутаций в гене Prnp, способствующих образованию PrPSc из PrPC (наследственные формы прионных заболеваний). [9,10]
Таким образом у прионных болезней может быть причиной генетический дефект, заражение извне или их комбинация.
Несмотря на ярые атаки критиков этой теории, сейчас практически все соглашаются с тем, что Прузинер был прав, и этому есть значительный объем экспериментальных подтверждений. К примеру, если представить, что воспроизведение PrPSc после попадания в организм происходит путем передачи патологической конформации на PrPC, то организмы, лишенные PrPC, должны быть устойчивы к прионной инфекции. Такой эксперимент провели с использованием трансгенных мышей, гомозиготных по делеции гена Prnp (Prnp0/0). Введение растертой ткани мозга мышей, больных скрепи, трансгенным мышам Prnp0/0 не приводило к развитию болезни ввиду отсутствия нормального PrP. Более того, оказалось, что в отсутствие PrPC не происходит не только воспроизведения приона, но и повреждения нервной ткани.
Окончательное доказательство концепции прионов долгое время сдерживалось невозможностью получения значительного количества PrPres – формы PrPSc, образуемой in vitro, которая устойчива к частичному протеолизу и способна вызывать болезнь при введении экспериментальным животным. Недавно было показано, что фрагмент рекомбинантного PrP мыши, синтезированный в Escherichia coli, образует фибриллы in vitro, которые при введении трансгенным мышам, экспрессирующим этот же фрагмент PrP, приводят к развитию прионного заболевания. [13]
Также в недавнее время была разработана система циклической амплификации прионной формы белка PrP, с помощью которой возможно формирование значительтного количества PrPres (искусственной патологической версии приона) in vitro. Это позволило получить и продемонстрировать инфекционность искусственно синтезированного приона.
Внимательный читатель заметит, что прионы, образующиеся в мозге овцы, вряд ли будут патогенны по отношению к человеку. И будут почти правы. Известно, что передача прионной инфекции между видами млекопитающих ограничена межвидовыми барьерами. Например, болезнь Крейцфельда-Якоба передается от человека человеку, и от человека шимпанзе; скрепи же передается среди овец и коз, но не передается шимпанзе. В то же время межвидовые барьеры не абсолютны. Межвидовые барьеры могут выражаться не столько в невозможности передачи инфекции животным отдаленного вида, сколько в удлинении инкубационного периода, а также в том, что заболевают не все, а какая-то часть экспериментально зараженных животных. Считается, что межвидовые барьеры вызваны различиями в первичной структуре PrP и модификациях у млекопитающих разных видов. Подтверждением этому послужили следующие наблюдения. Трансгенные мыши, экспрессирующие PrP хомяка, оказались высокочувствительны к заражению прионами хомяка в отличие от мышей дикого типа. Передача болезни Крейцфельда-Якоба от человека к мыши ограничена межвидовым барьером, однако трансгенные мыши, экспрессирующие PrP человека, подвержены заражению этой болезнью.
Также до сих пор встречаются трудности по заражению животных с помощью чистого прионного белка. Эти трудности можно легко объяснить.
Первая причина в том, что в клетках обычного организма белки подвергаются посттрансляционной модификации, которую достаточно трудно воспроизвести в экспериментальных условиях.
Вторая причина в том, что прион является белком мембранным и следует предполагать, что и структура его наиболее стабильна в условиях мембранно-подобного окружения, что и было показано недавними исследованиями.
В них было показано, что прионы в присутствии холестерина и фосфатидилэтаноламина гораздо легче образовывали патогенную форму и обладали гораздо большей инфекционностью.
Прионы — биологическое оружие
На этом можно было бы и закончить рассказывать про страшные прионы. Однако читатель законно спросит: « Причем же здесь биологическое оружие? Ведь чтобы произошло заражение необходимо, чтобы патогенная молекула приона попала в головной мозг. Не будем же мы сами себе делать трепанацию черепа».
На самом деле ситуация с возможными путями заражения оказалась гораздо хуже, чем можно было себе представить. В 1974 году был описан первый случай ятрогенного (из-за внешнего воздействия) заболевания болезнью Крейтцфельда-Якоба, обычно считавшейся генетической патологией.
Имеются описания 3 случаев передачи БКЯ в результате переливания крови от донора, у которого был диагностирован БКЯ во время вспышки этого заболевания в Великобритании [28]. От чего же произошла эта вспышка… Как обычно из-за жадности. БКЯ развилась у людей после употребления в пищу говядины, зараженной прионами.
В 1986 г. в Великобритании вспыхнула эпидемия заболевания прионнной болезнью у коров, также названной «коровьим бешенством», которая привела к гибели более чем 160 000 голов крупного рогатого скота [29]. Причиной было использование пищевых добавок мясокостной муки, когда из-за слабо контролируемых правил переработки побочных продуктов животного происхождения PrPSc от зараженных скрепи овец и другого крупного рогатого скота, попадал в корм для коров. Обычно в технологию получения такой муки после процессов тщательного измельчения исходного сырья включена обработка активными жирорастворителями, а также термообработка при температуре 130 оС. Однако в конце 70-х годов предприниматели, решив повысить питательную ценность мясокостной муки, снизили режим термообработки до 110 оС, а также уменьшили количество веществ, экстрагирующих жир. Именно эти изменения способствовали появлению и развитию эпидемии среди поголовья крупного рогатого скота.
Доказано, что эпидемия у коров привела к появлению нового типа БКЯ, получившего название «вариант БКЯ» [15]. Первые случаи вБКЯ были зарегистрированы в 1995 г., когда заболевание диагностировали у 2 британских подростков [16,17]. Из-за длительного инкубационного периода связь между заболеванием и зараженным мясом в Великобритании не была установлена до тех пор, пока заболеваемость у коров не переросла в эпидемию. Эпидемия была взята под контроль после массивного убоя скота и изменений в технологии производства, которые резко сократили загрязнение мяса компонентами нервной ткани. В Великобритании ежегодное число новых случаев вБКЯ, которое достигло пика в 2000 г., неуклонно снижается, и в 2013 г. был подтвержден только 1 случай заболевания [18].
У всех пациентов БКЯ развился после употребления в пищу мяса, полученного от заболевшего крупного рогатого скота. Но, несмотря на широкое распространение эпидемии, поразившей сотни тысяч голов крупного рогатого скота, относительно у немногих людей, которые употребляли в пищу мясо больных животных, развился БКЯ [33]. (вспомним про межвидовой барьер).
Инкубационный период (время между употреблением в пищу зараженной говядины и манифестацией симптомов) был длительным: большинство пациентов были заражены в конце 80-х годов, а пик заболеваемости пришелся на начало 2000-х, т. е. инкубационный период составил 11—12 лет. В последних диагностированных случаях инкубационный период достигал от 12 до более 20 лет [18.19].
Клинические проявления варианта БКЯ имеют отличия от других форм БКЯ. Болезнь настигает молодых людей в возрасте в среднем до 30 лет, ее начало характеризуется изменениями личности: больной утрачивает прежние интересы, начинает сторониться близких людей, у него развиваются тревожное состояние, бессонница, депрессия. Двигательные нарушения проявляются примерно через полгода после начала заболевания. Слабоумие наступает позднее, чем при классической форме, пациент осознает свое ухудшающееся состояние. Довольно быстро он теряет способность самообслуживания. Для вБКЯ типичны не только начало в более молодом возрасте, но и средняя выживаемость, превышающая 14 мес [18,19]. Вероятно, что различия в выживаемости между классической БКЯ и ее вариантом отчасти связаны с молодым возрастом пациентов.
Вот так, сама природа нам продемонстрировала возможность использования прионов как оружия с отсроченным сроком воздействия.
К глубочайшему сожалению в 2011 году во время экспериментальных работ по изучению болезни Крейтцфельда – Якоба на мышах была показана возможность воздушно – капельного заражения аэрозолями, содержащими прионные частицы.
Прионы – идеальное биологическое оружие?
В чем же заключаются главные достоинства:
1. Болезнь проявляется в отсроченном периоде, у злоумышленника есть время чтобы заразить как можно большее число людей. При этом все окружающие будут находится в абсолютном неведении.
2. Можно прекратить заражение, также незаметно, как и начать его. Найти следы и источник заражения спустя 5-7 лет будет невероятно трудно. Тем более нужно будет знать, что искать.
3. Высокая инфекционность прионов. При неудачном стечении обстоятельств для заражения теоретически достаточно одной молекулы неправильно свернутого белка, если эта молекула проникнет через гематоэнцефалический барьер и свяжется с обычной версией прионного белка на поверхности нейрона
4. Белок в обычных условиях присутствует в животном биоматериале и невозможно простыми способами отличить обычный прион от патогенного.
5. Прионы устойчивы во внешней среде, не разрушаются при стерилизации. Очень трудно расщепляются протеиназами. Способны связываться с частицами почвы, оставаясь стабильными долгое время.
6. Нет достаточно надежного диагностического теста для ранней стадии развития заболевания.
7. Против прионов нет вакцины или иного лекарства. Человек, заразившийся прионами обречен [9]
Помогаем террористам осуществить хитрый замысел
Прионы — это идеальная технология для террора. Существует и хорошо описана технология синтеза патогенных форм прионных белков[13].
Даже если представить, что обычному террористу сложно будет организовать биохимическую лабораторию, то бесчисленные стада животных никто не мешает использовать для получения большого количества мозгового вещества, зараженного БКЯ.
Никто и ничто не помешает террористам начать массовый синтез прионных белков и их добавление к сухому молоку, детским молочным смесям, мясному фаршу, мясо-костным субпродуктам, соевому шроту или любой другой субстанции, завод по производству которой окажется в зоне их досягаемости.
Если же представить, что в руки террористов попадет насильственным способом или по финансовым, идейным, иным соображениям талантливый биохимик, то никто не помешает ему синтезировать липидно-белковый аэрозоль с прионными частицами. Потом распылять ничем не определяемый аэрозоль в системах вентиляции. Этот способ более страшный, чем через пищу, так-как у носоглоточного узла есть тесная связь с мозгом и вероятность заражения увеличивается многократно.
Представим себе последствия заражения. Спустя 3-7, а может и все 15 лет на неограниченной территории начинается массовое развитие прионной болезни мозга. Паника, ужас, страх, разрушение. Целые города людей — зомби, чей мозг в буквальном смысле превращается в губку. Нет лекарства, нет надежды, только ужас неотвратимой скорой гибели.
Заключение
Использование такого оружия – только дело времени. Поэтому уже сейчас нужно предпринимать ряд шагов:
1. Проводить исследования по созданию надежных систем обнаружения патологических прионов в продуктах питания, воде, воздухе и делать этот тест обязательным к использованию на всей территории земного шара. Внедрять системы обнаружения прионных белков.
2. Искать возможность диагностики у человека появления патологических прионов. Тут есть радостные вести, что разработан высокочувствительный метод обнаружения патологических прионов. [12]
3. Искать способ излечения человека. Что представляется невероятно трудным, несмотря на многообещающие достижения с антиприонными антителами, способными проникать через гематоэнцефалический барьер.
4. Отслеживать на уровне спецслужб массовые случаи заражения животных прионными заболеваниями.
Новогоднее пожелание
Пожелаю никогда не встретить ни одной молекулы PrPSC!
Сам пока поищу возможность достать высокочувствительную диагностическую тест — систему, чтобы определить, не успели ли нас уже заразить…
Ссылки на источники
1. www.nobelprize.org/prizes/medicine/1976/gajdusek/biographical
2. Gajdusek, D. C.; Zigas, V. (1957-11-14). «Degenerative Disease of the Central Nervous System in New Guinea». New England Journal of Medicine. 257 (20): 974–978.
3. Hussain Khan, C.G. Bio-medical Paradigm // Bio-social issues in health. General editor, R.K. Pathak. New Delhi: Northern Book Centre, 2008. — p. 15
4. Chandler R.L. (1961) Lancet, 1,1378–1379
5. Alper T., Cramp W.A., Haig D.A., and Clarke M.C. (1967) Nature, 214, 764–766.
6. Griffith J.S. (1967) Nature, 215,1043–1044.
7. Chesebro B., Race R., Wehrly K., Nishio J., Bloom M., Lechner D., Bergstrom S., Robbins K., Mayer L., Keith J.M., et al. (1985) Nature,315, 331–333.
8. Prusiner S.B. (1982) Science, 216,136–144
9. Prusiner S.B. (1991) Science, 252,1515–1522
10. Prusiner S.B. (1993) Proc. Natl. Acad. Sci. USA Vol. 90, pp. 10962-10966, December 1993 Biochemistry
11. Saima Zafar et al., Handbook of Clinical Neurology, Vol. 165, 2019 (3rd series)
12. Serena Singh, Mari L. DeMarco JALM, January 2020
13. Nature Communications (2018) Chae Kim, Xiangzhu Xiao, Shugui Chen, Tracy Haldiman, Vitautas Smirnovas, Diane Kofskey, Miriam Warren, Krystyna Surewicz, Nicholas R. Maurer, Qingzhong Kong, Witold Surewicz & Jiri G. Safar Artificial strain of human prions created in vitro volume 9, Article number: 2166
14. Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, Will RG. Possible transmission of variant Creutzfeldta Jakob disease by blood transfusion. Lancet. 2004;363:417-421.
15. Collinge J. Human prion diseases and bovine spongiform encephalopathy (BSE). Hum Mol Genet. 1997;6(10):1699-1705
16. Bateman D, Hilton D, Love S, Zeidler M, Beck J, Collinge J. Sporadic Creutzfeldt Jakob disease in a 18-year-old in the UK. Lancet. 1995; 346(8983):1155-1156.
17. Britton TC, al-Sarraj S, Shaw C, Campbell T, Collinge J. Sporadic Creutzfeldt—Jakob disease in a 16-year-old in the UK. Lancet. 1995; 346(8983): 1155.
18. Soomro S, Mohan Ch. Biomarkers for sporadic Creutzfeldt Jakob disease. Annals of Clinical and Translational Neurology. 2016;3(6):465-472.
19. Imran M, Mahmood S. An overview of human prion diseases. Virol J. 2011;8:559.


