Существует много методов решения этой задачи, большинство из них относится к хроматографии. О том, как эти методы работают читайте под катом.

Неподвижная фаза «ловит» проплывающие мимо молекулы за их (-ОН)-группы.
Итак, мы получили нужный нам штамм и нарастили биомассу. Теперь нам нужно как-то вынуть из неё наш белок.
Первым делом мы берём нашу культуру и разливаем по флаконам для центрифугирования. Бактерии — довольно крупные и тяжёлые объекты, поэтому для их осаждения не нужны монструозные значения перегрузки при центрифугировании: 10000g в течение 20 минут хватит с лихвой. В итоге мы получим осадок на дне флаконов и жидкость (её называют супернатант). Жидкость мы выливаем, так как наш белок находится в бактериях, которые в осадке. Дальше можно действовать по обстоятельствам: либо сразу работать с полученным осадком, либо заморозить и хранить до тех пор, пока он не понадобится. Храниться он может очень долго. Дальнейшая процедура работы с осадком не зависит от того хранили мы его в морозильнике или нет.
Допустим, пришло время работать с осадком. Что дальше?
Первым делом нам нужно разрушить клетки, чтобы белок мог выбраться наружу. Для этого используют следующие способы:
- Последовательная заморозка-разморозка. Не самое эффективное средство для разрушения клеточной оболочки, зато оно безопасно для белка, поэтому часто начинают с нескольких таких циклов, а затем переходят к более эффективным методам;
- Обработка ультразвуком (соникация). Метод широко используется, он прост и эффективен. Обычно ультразвук подаётся напрямую в объём воды с помощью продолговатого «динамика», который вставляется в жидкость с клетками так, что конец «динамика» почти достаёт до дна. Это довольно «жёсткий» метод». Также есть более «мягкая» альтернатива — ультразвук подаётся в водяную ванночку, в которой плавают пробирки.

Выпускник MIT предупреждает — внимательно следите за своими соникаторами.
При проведении ультразвукового разрушения клеток важно следить за температурой: вода быстро нагревается под воздействием ультразвука, а нам совсем не нужна денатурация нашего белка и тем более нам не хотелось бы его кипятить. Поэтому в первом случае (с «вытянутой колонкой») пробирку с клетками вставляют в чашку со льдом и водой, причём льда должно быть много. Вода в данном случае выступает в качестве термоинтерфейса между льдом и охлаждаемой им пробиркой. В случае с «мягкой альтернативой» лёд добавляют непосредственно в водяную ванночку.

Демонстрация «жёсткой» ультразвуковой обработки.

Демонстрация «мягкой» ультразвуковой обработки.
- Добавление лизирующих агентов в среду. В основном это детергенты, то есть «мыло» (вообще слово «detergent» в английском означает «моющее средство»). Детергенты по сути являются родственниками липидов в мембранах клеток. Попадая в среду с клетками детергенты начинают встраиваться в мембраны, что приводит к их разрушению. А нам только этого и надо. Иногда также добавляют лизоцим — фермент, разрушающий клеточную стенку бактерий.
- Френч пресс. Суспензия клеток засасывается через входной клапан в рабочий цилиндр, откуда выдавливается прессом через узкую щель. Поперечные силы, возникающие при падении давления до атмосферного при прохождении клетками щели, приводят к разрушению клеточной стенки и мембраны.
По своему опыту могу сказать, что соникации в сочетании с детергентами обычно более чем достаточно.
Итак, мы получили клеточный лизат — «суп», состоящий из нужного нам белка и огромного количества «мусора» (хромосомной и плазмидной ДНК, кусков клеточной стенки, разнообразных белков, etc.). Как от всего этого избавляться?
Первый вопрос, на который нужно при этом ответить: «В каком виде наш белок существовал в клетке?». Тут есть два основных варианта:
- Белок был растворим до лизиса и с очень высокой долей вероятности таким и остался после него, значит мы не сможем осадить его центрифугированием. Зато мы можем осадить крупный «мусор», оставшийся после лизиса: так мы избавимся от кусков клеточной стенки, хромосомной ДНК и прочих крупных фрагментов. Осадок выбрасывается, а супернатант отправляется на хроматографию;
- Белок в условиях бактериальной цитоплазмы был нерастворим и он образовал т.н. «тельца включения», сохранившиеся после лизиса. Тельца включения — это белковые шарики, которые образуются в том случае, если белок во время синтеза не успевает сфолдироваться (то есть принять свою нормальную растворимую форму) до того, как столкнётся с другим таким же несфолдированным белком. У несфолдированных белков почти всегда есть торчащие наружу гидрофобные участки, с помощью которых они и прилипнут друг к другу. После того, как два белка «слиплись» у них уже недостаточно степеней свободы для того, чтобы завершить свой фолдинг. Потом к ним подплывает третий, четвёртый, пятый…. В итоге образуется белковая гранула. Обычно это происходит тогда, когда мы синтезируем в бактерии эукариотические белки (как я уже писал ранее, бактерия не умеет производить их фолдинг). В итоге мы имеем массу белка с «плохой» пространственной структурой. Но нет худа без добра, ведь в итоге образуются гранулы, почти полностью состоящие из нужного нам белка!
Поскольку в данном случае наш белок нерастворим, то при центрифугировании он вместе с «мусором» оказывается в осадке. Супернатант мы выбрасываем, а затем находящиеся в осадке тельца отмываются от всего ненужного следующим образом:
- Налили отмывочный буфер №1, в котором растворяется «мусорный компонент осадка №1»;
- Размешали осадок до однородности и поставили на шейкер (прибор, который взбалтывает);
- Отцентрифугировали и вылили супернатант. В итоге то, что растворилось в отмывочном буфере №1 мы отделили от осадка;
- Налили отмывочный буфер №2, в котором растворяется «мусорный компонент осадка №2»;
- Ну и так далее. В общей сложности обычно нужно около 5 промывок.
Ещё раз напомню, что если мы имеем дело в с тельцами включения, то мы имеем дело с несфолдированным белком, а значит он не в состоянии выполнять заложенные в него функции. Поэтому перед последующей очисткой проводят его рефолдинг, то есть приводят в активную форму. Самый распространённый метод рефолдинга состоит из следующих этапов:
- Растворить тельца в «нефизиологических» условиях. Обычно тельца хорошо растворяются при высоких значениях рН (около 12) в присутствии двухмолярной мочевины. Идея этого метода в том, что при высоком рН в среде очень мало протонов и все, кто мог бы от них избавиться с лёгкостью это делают. Протон заряжен положительно, значит все доноры протонов получают отрицательный заряд, ну а дальше всё делает закон Кулона — имеющие большой отрицательный заряд белки начинают отталкиваться друг от друга;
- По капле добавлять весь объём «раствора белка» из первого пункта в гораздо больший активно перемешиваемый объём буфера с комфортными для белка условиями. Идея этого этапа в том, что при низкой концентрации белка у него гораздо больше шансов сфолдироваться перед тем, как он встретится со своим несфолдированным собратом. Обычно покапельный перенос белкового раствора производят в объёмы буфера, превышающие объём самого раствора минимум в 10 раз.
Итак, у нас готовы некоторые объёмы растворов белка в сфолдированном состоянии. Дальнейшая процедура одинакова для любых «начальных условий» — мы фильтруем наши растворы через фильтры с размером пор 100-450 нм (чтобы не засорить хроматографические колонки и сам хроматограф), а затем проводим хроматографию.
Типы хроматографии
Принцип любой хроматографии заключается в том, что различные компоненты раствора двигаются через колонку с разными скоростями, в итоге шедшие «на входе в колонку» вместе компоненты оказываются на выходе в разное время.
Но сначала вспомним ещё один метод — белковый электрофорез в геле.
Этот метод позволяет разделять белки согласно их массам. Для этого в пробу добавляют буфер, содержащий SDS и краситель (обычно это бромфеноловый синий). При этом плотность буфера выше плотности воды, это сделано для того, чтобы смесь образца и буфера было проще вносить в лунки на геле. Краситель добавляют для контроля процедуры внесения образца в лунку, а также он служит в качестве индикатора того, что форез пока заканчивать: краска движется быстрее белка, поэтому когда она достигает конца геля форез прекращают. Это очень удобно, так как движение белка по гелю мы не видим.

SDS-ПААГ электрофорез. Лучше один раз прочитать и увидеть, чем сто раз прочитать.
SDS является ключевым компонентом: этот отрицательно заряженный детергент (ПАВ) денатурирует белки, после чего он их «окружает», при этом получается, что количество прилипших к белку молекул SDS прямо пропорционально его массе. Тогда из закона Кулона следует, что при отсутствии какого-либо сопротивления движению все окружённые молекулами SDS белки имели бы одинаковые ускорения в электрическом поле (сила растёт линейно с ростом заряда, но линейно с зарядом растёт и масса).
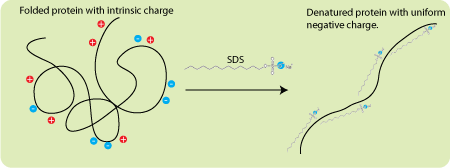
Схема действия отрицательного заряженного детергента SDS на белок. Сначала он его денатурирует, а затем «обволакивает». В итоге белок заключается в состоящую из SDS отрицательно заряженную оболочку, заряд которой прямо пропорционален массе белка.
И тут нам на помощь приходит ПААГ (ПААГ — полиакриламидный гель). При полимеризации он образует сеть каналов приблизительно одного и того же диаметра, через которую при прочих равных маленькие объекты движутся быстрее больших. Очевидно, что тяжёлый белок крупнее лёгкого, поэтому лёгкие белки двигаются быстрее.
После окончания процедуры электрофореза гель окрашивают. Точнее, окрашиваются те места, в которых в геле находится белок — получаются синие полоски (или чёрные, смотря чем красить). Для того, чтобы понять какой массе соответствует та или иная полоса нужно с чем-то сравнивать, поэтому на каждый гель наносят т.н. «маркер молекулярной массы» — смесь компонентов с известными массами.
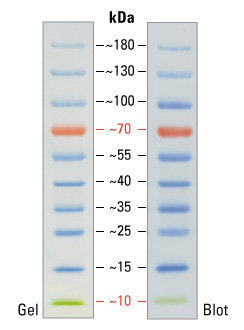
Пример полосок маркера молекулярной массы. Данный маркер удобен ещё и тем, что две полосы у него окрашены в индивидуальный цвет: красная (70 килодальтон) и зелёная (10 килодальтон). Это очень помогает экспериментатору ориентироваться в самом маркере. 1 дальтон, он же атомная единица массы, определяется как 1⁄12 массы свободного покоящегося атома углерода-12, находящегося в основном состоянии.
Казалось бы, чудо, а не метод — полоски не перекрываются, а значит белки идеально разделились! Но на деле учёные так и не научились проводить белковый электрофорез в промышленных масштабах и поэтому мы до сих пор используем колоночную хроматографию.
Примерная схема любой хроматографии. Колонка представляет собой чем-то наполненный цилиндр. Это «что-то» называется «неподвижная фаза», а «подвижной фазой» является раствор, компоненты которого нам нужно разделить. Компоненты движутся с разной скоростью и потому выходят из колонки порознь.
Большинство хроматографических приёмов основаны на том, что проплывающие мимо белки цепляются за неподвижный наполнитель колонки «с разным энтузиазмом»: те, кто «цепляются» часто и сильно движутся медленнее всех, а те, кто не «цепляется» ни за что быстро покидают колонку. В итоге существенная часть смеси часто выходит из колонки, так и не «зацепившись» за неё — они выходят вместе в самом начале хроматографии (эта фракция называется «проскок», так как они проскочили колонку, не «зацепившись» за неё). Но что делать с теми, кто «зацепился»?
Для начала разберёмся с тем, что происходит в колонке с теми, кто «цепляется». Сам «зацеп» происходит потому, что на поверхности неподвижной фазы есть что-то такое, что способно на время «захватить» проплывающий мимо белок (о том, как это происходит — чуть ниже). То есть «цепляющийся» белок проводит некоторое время в связанном с твёрдой фазой состоянии, затем отрывается от ней (в силу собственного теплового движения, соударений и т.д.), плывёт дальше, опять связывается с твёрдой фазой, опять отрывается и так до тех пор, пока он не покинет колонку. Понятно, что чем чаще и чем крепче он связывается с неподвижной фазой, тем медленнее он движется вдоль колонки.
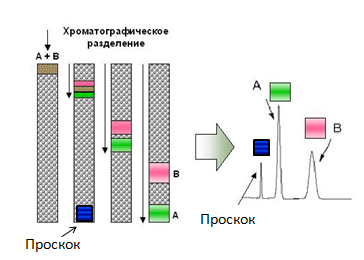
Принцип хроматографии. То, что не связывается с колонкой (проскок) движется со скоростью подвижной фазы (с поправкой на размер). Связывающиеся компоненты имеют разную скорость, что и приводит к их разделению на выходе.
Вообще говоря, только в случае большого невезения в смеси будут разные компоненты, которые движутся с одинаковой скоростью, поэтому мы можем просто подождать, когда они выйдут из колонки. Но зачастую «просто ждать» очень долго, так как движутся компоненты медленно. В этом случае их нужно «подгонять», а способ их «подгонять» зависит от типа колонки.
Наконец, рассмотрим сами хроматографические методы, которые подходят для промышленного получения белов.
- Ионообменная хроматография. Идея метода в том, что неподвижная фаза в условиях проведения хроматографии имеет заряд. Если заряд положительный, то говорят об «анионообменной хроматографии», если отрицательный, то о «катионообменной».
Логика названия проста, рассмотрим её на примере анионообменника: к положительно заряженной твёрдой фазе колонки «липнут» отрицательно заряженные компоненты смеси. При этом если мы будем следить за какой-то конкретной положительно заряженной группой на поверхности твёрдой фазы, то к ней поочерёдно будут «липнуть» и отрываться те или иные отрицательно заряженные компоненты. Таким образом, происходит своего рода обмен анионами между неподвижной и подвижной фазами, отсюда и название — «анионообменная хроматография».
Очевидно, что если мы ходит использовать анионообменную хроматографию, то мы должны проводить разделение смеси в таких условиях, при которых твёрдая фаза заряжена положительно. При этом у нас есть на выбор два варианта дальнейшего развития событий в зависимости от того, какой заряд при этих условиях имеет белок: если «-«, то он «сядет» на колонку, а если «+», то он окажется в проскоке.
Как ускорить выход связанных компонентов, то есть как провести «элюцию»? Можно действовать двумя способами:
- Можно изменить заряд неподвижной фазы и связавшихся компонентов с помощью изменения рН подвижной фазы. Важно производить процесс изменения рН постепенно, тогда компоненты ускорят своё движение, но не «вывалятся» при этом все сразу;
- Добавить в колонку другие анионы. Действительно, колонка располагает конечным числом мест, за которые могут зацепиться анионы и если мы чем-то займём все эти места, то компоненты смеси будут двигаться быстрее. Для анионообменной хроматографии типично добавление анионов хлора. Внесение конкурирующих анионов также следует проводить постепенно.
Методика проведения катионообменной хроматографии в целом та же, только всё наоборот.
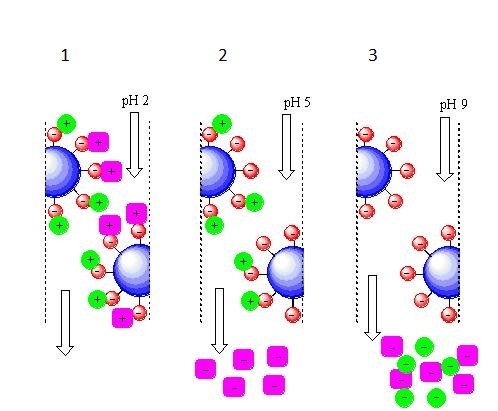
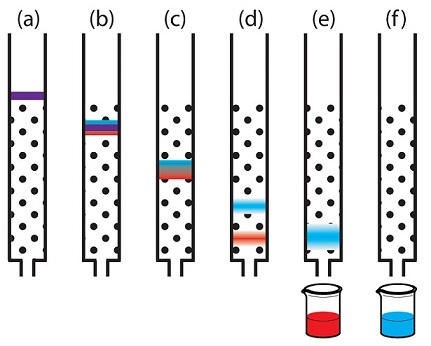
Катионообменной хроматография с элюцией за счёт изменения рН подвижной фазы.
1 — при рН=2 в подвижной фазе очень много протонов, поэтому все компоненты смеси нахватались их из жидкости, получив при этом положительный заряд;
2 — при рН=5 количество протонов в подвижной фазе снизилось и «пурпурный» компонент уже выступает не в качестве акцептора, а в качестве донора протонов, стало быть у него при данном значении рН заряд отрицательный и он покидает колонку. «Зелёный» компонент всё ещё выступает в роли акцептора протонов и он сохраняет положительный заряд при данном значении рН, значит он пока держится за колонку;
3 — при рН=9 в подвижной фазе очень мало протонов, «зелёный» компонент тоже становится донором протонов, приобретает отрицательный заряд и покидает колонку последним.
В общем и целом выбор типа ионообменника и условий проведения разделения зависит от того, что мы хотим выделить и что ещё есть в нашей смеси.
- Ещё один метод разделения основан на гидрофобности (~неполярности) и гидрофильности (~полярности) тех или иных веществ. Идея тут точно такая же, как и в случае с ионообменником: компоненты согласно своим физическим свойствам «мечутся» между гидрофобной и гидрофильной средами, кто-то задерживается больше, кто-то меньше.
Прежде всего напомню, что гидрофобное растворяется в гидрофобном, а гидрофильное в гидрофильном. Зачастую «гидрофобный» и «неполярный» это одно и то же, как и «гидрофильный» и «полярный», хотя в общем случае это не так. Примером неполярного растворителя является этаном а примером полярного — вода.

Полярный медведь сетует на то, что он растворяется в воде, которая является полярным растворителем. «Неполярному» бурому медведю это не грозит.
Теперь рассмотрим пример. Допустим, неподвижная фаза гидрофобна (неполярна). Тогда подвижная должна быть в целом гидрофильной (полярной). Все гидрофобные компоненты «сядут» на колонку, так как плавать в гидрофильной подвижной фазе им «страшно». Гидрофильные же компоненты окажутся в проскоке, так как им гидрофильная подвижная фаза по душе. Затем «севшие» на колонку гидрофобные компоненты нужно элюировать постепенным увеличением гидрофобности подвижной фазы. Регуляция гидрофобности подвижной фазы осуществляется за счёт насоса, который качает одновременно полярный и неполярный растворитель из двух разных резервуаров через колонку. От соотношения между ними и зависит итоговая полярность подвижной фазы.
- Металлохелатная хроматография с гистидиновым тагом (His-tag). Метод основан на том факте, что участки из шести гистидинов (гистидин — одна из 20 протеиногенных аминокислот) хорошо связываются с находящимися на колонке ионами никеля через свои имидазольные группы. Метод удобен тем, что мало в каком природном белке есть гистидиновый таг, значит с высокой долей вероятности все белки, кроме нашего будут в проскоке.
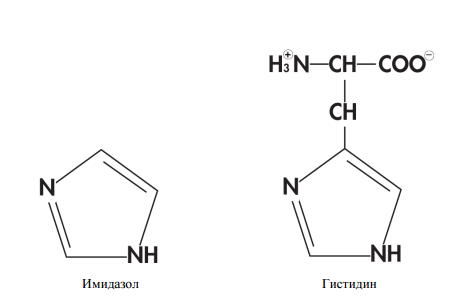
Строение молекул имидазола и гистидина.
Минус метода в том, что нужно модифицировать белок, а это может сказаться на его свойствах. Добавление 6 гистидинов тага к белку происходит ещё на стадии создания гена, а чтобы минимизировать влияние тага на белок эти аминокислоты добавляют в начале или конце аминокислотной последовательности. Естественно, метод не сработает, если белок таков, что его начало и конец спрятаны внутри: ионы никеля и таг просто не смогут встретиться.
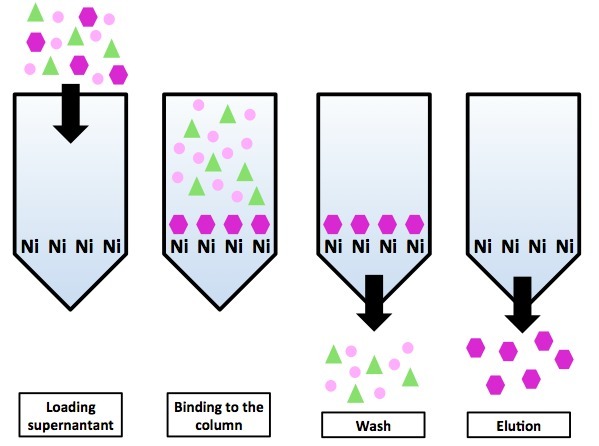
Схема хроматографии на никилевой колонке с использованием гистидинового тага. Все белки, кроме белка-«пурпурного шестиугольника» не имеют His-тага и оказываются в проскоке. Белки-«пурпурные шестиугольники» элюируются нанесением на колонку имидазола.
«Снять» белок с колонки можно постепенным добавлением имидазола. Суть метода элюции та же, что и у ионообменников — имидазол конкурирует с несущим His-таг белком за сайты связывания на неподвижной фазе (только на ионообменнике сайтами связывания были абстрактные ионы, а на металлохелатной колонке сайтами связывания являются ионы никеля). В итоге он занимает большинство доступных сайтов связывания и белку ничего другого не остаётся, кроме как покинуть колонку.
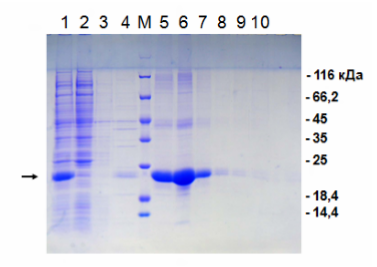
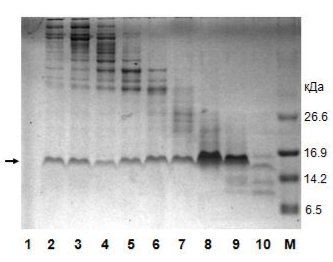
Электрофорез по итогам металлохелатной хроматографии на никеле.
1 — исходный «грязный» материал для нанесения на колонку; 2 — проскок; 3 и 4 — промывка без имидазола; М — маркер молекулярной массы; 5-10 — последовательная элюция имидазолом. Нужный нам белок помечен стрелкой слева. Как мы видим, в проскоке нет нашего белка, зато в нём есть много «мусора». Элюат же представляет собой очищенный целевой белок. Справа обозначены массы соответствующих полос маркера молекулярной массы.
Метод потенциально мог быть удобен ещё и и тем, что существует зависимость в силы связывания от количества гистидиновых тагов в составе белка. Таким образом, можно было бы пришивать к одному белку таг только в начале, а к другому с обоих концов и синтезировать их в одной бактерии. Но на деле это не очень практично: разница в концентрациях имидазола, которые элюируют такие белки невелика (примерно 75 мМ и 100 мМ имидазола для одного и двух тагов, соответственно), значит существует вероятность возникновения сложностей с их разделением из одной смеси. А сделать два отдельных штамма это ничуть не сложнее, чем сделать один с двумя генами.
Другой возможной трудностью при работе с His-тагом является существующая в некоторых экспериментах необходимость в получении белка без каких-либо модификаций. В принципе отрезать ферментами таг от белка возможно, но часть белка при этом всё таки останется с тагом (то есть «КПД < 1»), также это приведёт к тому, что нам нужно будет проводить дополнительную стадию очистки от этих самых ферментов. При этом каждая стадия очистки сопряжена с потерями целевого продукта.
- Гель-фильтрация. Сразу оговорюсь, что данный метод можно назвать подходящим для выделения белков лишь условно: если нам нужно получить небольшое количество белка для исследований, то использование гель-фильтрации имеет смысл, но эта методика не годится для работы с действительно промышленными объёмами, и вот почему. Во всех предыдущих методах скорость движения связавшихся с колонкой компонентов в отсутствии «подгоняющих факторов» настолько мала, что её можно вообще считать близкой к нолю.
С гель-фильтрацией всё обстоит иначе: все компоненты, даже самые крупные, движутся со вполне заметными скоростями. Дело в том, что гель-фильтрация не использует какие-то специфические взаимодействия, как это было с предыдущими типами хроматографии (взаимодействие зарядов в ионообменниках, гидрофобные взаимодействия и узнавание ионов никеля имидазольными группами гистидина), а именно благодаря ним и обеспечивалось эффективное удержание некоторых компонентов. Это приводит к существованию сверхсущественного лимитирующего фактора: мы не можем наносить на гель-фильтрацию образец в объёме более 1-2% от объёма колонки. Действительно, околонулевая скорость компонентов в предыдущих методах позволяла не бояться того, что они размажутся даже при длительном нанесении, поэтому на колонку объёмом 10 мл запросто можно нанести смесь объёмом 400 мл: всё, что связалось будет сидеть в самом начале колонки вплоть до начала элюции. При гель-фильтрации же ни один компонент околонулевой скорости не имеет.
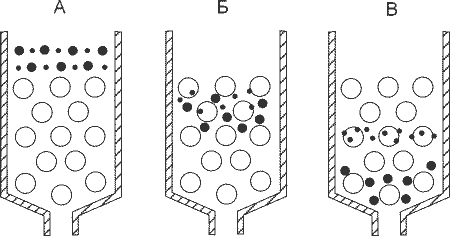
Принцип действия гель-фильтрации: крупные молекулы проходят только в крупные поры и поэтому движутся быстрее маленьких молекул, которые могут проникать в маленькие поры.
Как гель-фильтрация работает? Для её проведения нужна колонка, которая заполнена материалом, испещрённым каналами самого разного диаметра. При этом путь, состоящий из широких каналов является коротким, но если постоянно проникать в более мелкие канальцы и долго в них петлять, то путь заметно удлинится. Поэтому при гель-фильтрации крупные молекулы покидают колонку раньше, чем маленькие.
Электрофорез по итогам гель-фильтрации. Фракции пронумерованы в порядке выхода из колонки. Видно, что с каждой последующей фракцией масса белка возрастает. М — маркер. Справа обозначены массы соответствующих полос маркера молекулярной массы.
Выше было сказано про существенный недостаток гель-фильтрации, но есть и существенное преимущество. Механизм разделения полностью независим от того, что из себя представляет подвижная фаза, поэтому мы легко можем проводить элюцию в том буфере, который нам будет нужен в дальнейшем. Гель-фильтрация — единственный хроматографический метод, позволяющий произвести смену буфера на нужный нам в очень широких пределах.
Ну что же. Вот мы и выделили наш белок и теперь его можно использовать для структурных или функциональных исследований, в качестве реактива в научной лаборатории или для проведения экологических анализов, в медицинских целях или в промышленности.
Источник


